
Abstract
Coronavirus disease 2019 (COVID-19), resulting from infection by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), can cause severe and fatal pneumonia along with other life-threatening complications. The COVID-19 pandemic has taken a heavy toll on the healthcare system globally and has hit the economy hard in all affected countries. As a result, there is an unmet medical need for both the prevention and treatment of COVID-19 infection. Several herbal remedies have claimed to show promising clinical results, but the mechanisms of action are not clear. We set out to identify the anti-viral natural products of these herbal remedies that presumably inhibit the life cycle of SARS-CoV-2. Particularly we chose four key SARS-CoV-2 viral enzymes as targets: Papain-like protease, Main protease, RNA dependent RNA polymerase, and 2’-O-ribose methyltransferase, which were subjected to an unbiased in silico screening against a small molecule library of 33,765 compounds originating from herbs and medicinal plants. The small molecules were then ranked based on their free energy of fitting into the “druggable” pockets on the surface of each target protein. We have analyzed the best “fit” molecules and annotated them according to their plant sources and pharmacokinetic properties. Here we present a list of potential anti-viral ingredients of herbal remedies targeting SARS-CoV-2 and explore the potential mechanisms of action of these compounds as a framework for further development of chemoprophylaxis agents against COVID-19.
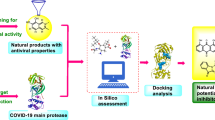
Graphic abstract

Similar content being viewed by others

1 Introduction
Coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This virus belongs to the enveloped RNA virus Coronaviridae family with close relation to the other two highly pathogenic members SARS-CoV and Middle East Respiratory Syndrome coronavirus (MERS-CoV), which were responsible for the worldwide outbreaks of respiratory diseases in 2002–2003 and 2012, respectively [1,2,3].
SARS-CoV-2 contains a single positive-sense RNA strand of approximately 26,000 to 32,000 bases. At the 5’ end, two overlapping ORFs, ORF1a and ORF1b, encode the gene for the replicase complex. The rest of the genome encodes ORFs for accessory proteins and structural proteins including the Spike (S), Envelope (E), Membrane (M), and nucleocapsid (N) proteins. Upon SARS-CoV-2 entry into the host cell, two polyproteins pp1a and pp1ab are translated from ORF1a and ORF1b, and subsequently cleaved into 16 non-structural proteins (NSPs 1–16) by two proteases, Papain-like protease (NSP3-PLpro), and Main Protease (NSP5-Mpro). Next, the membrane associated replication-transcription complex (RTCs) is assembled from the NSPs containing the key replication enzyme RNA-dependent RNA polymerase (NSP12-RdRp) to drive SARS-CoV-2 genome replication, subgenomic transcription, and structural protein synthesis. In addition, the SARS-CoV-2 genome encodes several RNA processing enzymes including NSP16 with 2’-O-ribose methyltransferase (NSP16-2OMT) activity involved in viral RNA 5’-capping and maturation. These evolutionary conserved viral enzymes are essential for virus replication and infectivity [1, 2, 4, 5]. Therefore, tremendous effort has been focused on understanding the structures and functions of SARS-CoV-2 NSPs in viral pathogenesis and targeting them for anti-viral therapy.
Different herbal remedies have been used as preventive recipes, nutritional supplement, and treatment approaches for COVID-19 in several Asian countries and have been shown to effectively reduce virus transmission and attenuate disease progression [6,7,8,9]. However, the mechanisms of action for the herbal remedies in anti-viral therapies are not understood. It is noteworthy that many herbs used in traditional Chinese medicine do contain active anti-viral compounds such as glycyrrhizin from liquorice root and have been widely used as key ingredients in recipes for treatment of influenza, hepatitis, and other viral infections [10,11,12]. Thus, the analyses of the anti-viral ingredients of herbal remedies would lead to more effective development of chemoprophylaxis for prevention and treatment of COVID-19 and other coronavirus infections.
Here, we report a comprehensive in silico screening and molecular docking study of four essential pathogenic proteins of SARS-CoV-2, including two proteases, NSP3-PLpro and NSP5-Mpro, and two proteins critical for RNA replication and processing, NSP12-RdRp and NSP16-2OMT, using a phytochemical compound library. The top candidates were evaluated and ranked based on their theoretical free energy upon binding to the targeted “druggable” sites. We identified several top candidates including Albanol B and Amentoflavone that were the active ingredients from medicinal herbs with previously shown antiviral effects. This study could serve as a framework for further development of bioactive antiviral natural compounds for COVID-19.
2 Methods
2.1 Database construction, protein preparation, and virtual screening
A database of 33,765 phytochemical compounds was constructed for structural-based virtual screening, which includes 8445 different species from herbs and medicinal plants used in traditional Chinese medicine according to the TCM Database@Taiwan [13], and was cross- referenced using compound ID from Zinc database or Pubchem [14]. Compound structures were converted to pdbqt format using Applied Chemistry Software Openbabel [15].
Virtual screening was performed using a computer-aided drug screening docking platform powered with Autodock Vina [16, 17]. The screening strategy is outlined in Figure S1 and involves two major steps. In the first step, potential “druggable” pockets were identified on the surface of the target proteins using the automatic surface shape-scanning program PocketPicker [18]. The second step performs computational docking and screening via MGLtools AutoDock Vina to identify the compounds that will bind to the “druggable” pockets. The docking program uses a scoring system to predict the best-bound conformation of the small molecules. The potential energy contributed by the protein–ligand intermodular interactions from the best bound conformations (bound free energy) were optimized to produce the global minimal potential energy of the protein–ligand complex [19, 20]. Four SARS-CoV-2 proteins, NSP3-PLpro, NSP5-Mpro, NSP12-RdRp, and NSP16-2OMT were chosen as target proteins. The individual targeted protein was prepared as described and the virtual screening process was validated for each protein using its complex structure bound with a ligand. After virtual screening, the best binding complexes for each target were selected for further analyses including visual inspection of potential ligand–protein interactions, molecular dynamics simulations and prediction of pharmacokinetic properties.
2.2 SARS-CoV-2 NSP3-PLpro
The SARS-CoV-2 core NSP3-PLpro sequence was derived from the NSP3 papain-like proteinase domain, downloaded from the NCBI protein database (protein ID: YP_009725299.1). The SARS-CoV NSP3-PLpro structure (PDB 5E6J_A) was used as template with the SWISS-MODEL online service (https://swissmodel.expasy.org/) to build the initial SARS-CoV-2 NSP3-PLpro structure model. The druggable pocket, predicted with PocketPicker using software PyMOL was located between the predicted ubiquitin binding site and the active site [18, 21]. The docking setup parameters include the following: center for grid box at x = 120, y = 8, z = 288; size for grid box at x = 22, y = 20, z = 18; num_modes = 9. The SARS-CoV-2 NSP3-PLpro structure PDB 7JRN in complex with GRL0617 was used to validate the modeling pocket and the docking process, and PDB 6XAA was used to validate the docking results for the candidate small molecules.
2.3 SARS-CoV-2 NSP5-Mpro
The SARS-CoV-2 NSP5-Mpro sequence was downloaded from the NCBI database (protein ID: YP_009725301.1). The SARS-CoV-2 NSP5-Mpro protein structure was downloaded from PDB database (PDB 6LU7_A). The druggable pocket was generated with PocketPicker using the PyMOL software. Virtual screening for NSP5-Mpro was performed with the setup parameters as the following: center for grid box at x = − 12, y = 15, z = 68; size for grid box at x = 20, y = 20, z = 22; num_modes = 9. The modeling pocket and the docking process was validated using SARS-CoV-2 NSP5-Mpro inhibitor N3 (PRD_002214), which was the bound ligand in PDB 6LU7.
2.4 SARS-CoV-2 NSP12-RdRp
SARS-CoV-2 NSP12-RdRp was downloaded from the NCBI protein database (protein ID: YP_009725307.1). The SARS-CoV NSP12 structure (PDB 6NUR_A) was initially used as the template with the SWISS-MODEL online service to build the SARS-CoV-2 NSP12-RdRp structure model. SARS-CoV-2 NSP12-RdRp ligand-binding pocket was generated with PocketPicker using software PyMOL. Virtual screening for NSP12-RdRp was performed with the setup parameters as the following: center for x = 142, y = 150, z = 155; size for grid box at x = 20, y = 24, z = 20; num_modes = 9. The SARS-CoV-2 NSP12-RdRp structure PDB 7BV2 in complex with Remdesivir was used to validate the modeling pocket and the docking process, and PDB 6M71 was then used to validate the docking results for the candidate small molecules.
2.5 SARS-CoV-2 NSP16-2OMT
The SARS-CoV-2 NSP16-2OMT sequence was downloaded from the NCBI database (protein ID: YP_009725311.1). The SARS-CoV NSP16 protein structure (PDB 3R24_A) was initially used to generate the SARS-CoV-2 NSP16-2OMT protein model with SWISS-MODEL online service. The SARS-CoV-2 NSP16-2OMT ligand-binding pocket was generated with PocketPicker using PyMOL. Two druggable pockets were used for virtual screening. The parameters for pocket one, the S-Adenosylmethionine binding site, were as the following: center for grid box at x = 55, y = 63, z = 65; size for grid box at x = 16, y = 18, z = 14; num_modes = 9. The parameters for pocket two, the RNA-cap-binding groove, were as the following: center for grid box at x = 63, y = 63, z = 55; size for grid box at x = 14, y = 18, z = 18; num_modes = 9. The SARS-CoV-2 NSP16-2OMT structure (PDB 6W4H) was used to validate the modeling pocket and the docking process using the bound ligand SAM, and followed by validation of the docking results for the selected small molecules.
2.6 Molecular dynamics (MD) simulation
Molecular dynamics simulation was used to clarify the dynamic behavior of the protein–ligand complexes at an atomic level. The protein–ligand complex structures from covalent docking were submitted to MD simulations using Gromacs 5.0.2 [22]. CHARMM force field for all atoms was chosen to run MD simulation for a time scale of 50 ns. All protein–ligand systems were solvated with three-point transferable intermolecular potential (TIP3P) and their charges were neutralized via adding Na or Cl ions. Energetic minimization of the protein–ligand systems was performed through the steepest descent algorithm at a tolerance value of 1000 kJ mol−1 nm−1, followed by the equilibration with position restraint on the protein molecules for 0.1 ns using NVT and NPT ensembles. Electrostatic interactions were evaluated by Particle Mesh Ewald summation [23].
2.7 ADME prediction
SwissADME was used to calculate the Absorption, Distribution, Metabolism, and Excretion (ADME) properties of the candidate compounds [24]. The molecule SMILES file was submitted to SwissADME (http://www.swissadme.ch) to calculate grouped parameters to evaluate physicochemical properties, lipophilicity, pharmacokinetics, and drug-likeness. The candidate compound physicochemical properties were represented by the chemical formula, Molecular Weight (MW), the counts of specific atoms (H-bond acceptors and H-bond donors), and Topological Polar Surface Area (TPSA). Lipophilicity was represented by the predicted partition coefficient between n-octanol and water (LOGPo/w). Water solubility was represented by LOGS, the decimal logarithm of the molar solubility in water. SwissADME also provided qualitative solubility classes (soluble, moderately soluble, poorly soluble, and insoluble) describing general water solubility of a particular compound. The pharmacokinetic properties were represented by the skin permeability coefficient (Kp), passive human gastrointestinal absorption, and blood–brain barrier (BBB) permeation. Other pharmacokinetic parameters included substrates or non-substrates of the permeability glycoprotein (P-gp) and cytochromes P450 five major isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4). Bioavailability was used to predict the oral bioavailability of a compound. Drug-likeness was used to assess the probability of a molecule to become an oral drug (0 to1, least probable to most probable), commonly evaluated by the Lipinski filter [19].
3 Results
We hypothesized that bioactive phytochemicals inhibit SARS-CoV-2 infectivity and/or viral replication in human cells through targeting one or more key enzymes: NSP3-PLpro, NSP5-Mpro, NSP12-RdRp, and NSP16-2OMT (Fig. 1) [25]. To test this, we performed an unbiased in silico screening against a library of 33,765 compounds originating from herbs and medicinal plants targeting each of these four SARS-CoV-2 proteins.

The SARS-CoV-2 proteins selected for virtual screening. Schematic presentation of the SARS-CoV-2 genome organization showing the four proteins included in this study, NSP3-PLpro (PDB 5E6J, 6XAA), NSP5-Mpro (PDB 6LU7). NSP12-RdRp (PDB 6NUR, 7BTF), and NSP16-2OMT (PDB 3R24, 6W4H). The druggable pockets chosen for in silico screening were indicated in ball-filling models and prepared using PyMOL
3.1 SARS-CoV-2 NSP3-PLpro
The NSP3-PLpro is one of the two essential proteases used by SARS-CoV-2 virus to generate NSP 1–4 as part of the viral replication complex from the initial polyproteins translated from viral RNA. NSP3-PLpro is a multifunctional enzyme, carrying viral protease activity as well as deubiquitinase and deISGylation activity for antagonization of the host cell antiviral immune response and promotion of viral replication [21, 26,27,28]. It is indispensable for SARS-CoV-2 infection and viral replication, thus making it an ideal target for screening of bioactive compounds.
The sequence alignment of SARS-CoV NSP3-PLpro and SARS-CoV-2 NSP3-PLpro (protein ID: YP_009725299.1) revealed that these two proteins share 83% sequence identity and over 90% similarity (Figure S2). The catalytic active site including the catalytic triad Cys112/His273/Asp287 (PDB 5E6J) and the substrate “LXGG” peptide recognition sequence are completely conserved (Figure S2) [27, 29]. Surface examination of SARS-CoV-2 NSP3-PLpro revealed an elongated and shallow groove located between the “Thumb” and “Palm” domain spanning the substrate “LXGG” peptide entry point with the finger domain on the left and the catalytic triad on the right (Fig. 2A). The substrate-binding loop (residues 382GNYQCG387), also known as the BL2 loop of SARS-CoV NSP3-PLpro (PDB 5E6J), is comprised of a β-turn, acting as a gatekeeper shaping the substrate-binding groove [29]. This groove was then selected for in silico screening attributing to its importance in NSP3-PLpro substrate recognition and cleavage, which has been targeted by a group of naphthalene inhibitors [27, 30, 31].

The potential inhibitors of SARS-CoV-2 NSP3-PLpro identified by in silico screening. A The substrate-binding groove located between the Thumb and Palm domain was selected as the druggable pocket for NSP3-PLpro in silico screening. The substrate-binding loop (BL2 loop) was depicted. B The 2D and 3D structure of the identified compound Glycobismine F as it was fitted in the binding pocket. The potential compound–protein interactions are depicted. C–E The 2D structures of three top-ranked compounds from SARS-CoV-2 NSP3-PLpro virtual screening Rheidin A, Albanol B, and Mahuangnin A
Among the top ranked candidate compounds, the small molecule Glycobismine F from the herb Glycosmis pentaphylla was identified, which fits the substrate-binding pocket with affinity-free energy of − 9.6 kcal/mol in the initial NSP3-PLpro model built by SARS-CoV and -9.6 kcal/mol using SARS-CoV-2 NSP3-PLpro (PDB 6XAA) (Fig. 2B). Glycobismine F occupies the same space as the previous identified NSP3-PLpro naphthalene inhibitor compound 24 (PDB 3E9S) and compound 3 k and 3j (PDB 4OW0 and 4OVZ). Glycobismine F was found to form multiple contacts with NSP3-PLpro including Tyr384 and Gln385 from the substrate-binding loop (BL2) by H-bond, π–π, and van der Waals interactions. To further predict the behavior of NSP3-PLpro binding by Glycobismine F, MD simulation was performed on NSP3-PLpro complexed with Glycobismine F for 50 ns (Fig. S5). Root Mean Square Deviation (RMSD) was calculated for NSP3-PLpro and Glycobismine F, respectively, over the course of MD simulation (Fig. S5E, F). MD analysis confirmed the stability of the protein–ligand complex, indicated by the low RMSD values observed for NSP3-PLpro and Glycobismine F around 0.2 nm throughout the course of the MD run. MD analysis further showed Glycobismine F interaction with the BL2 loop residues Tyr384 and Tyr389, occupying the same surface as the naphthalene inhibitors (Fig. S5). This particular space used by Glycobismine F and the naphthalene inhibitors were shown to be unique for SARS-CoV PLpro, as the naphthalene inhibitors that bound to SARS-CoV and SARS-CoV-2 NSP3-PLpro showed no or much lower inhibitory potency towards MERS-CoV PLpro and other viral proteases such as HCoV-NL63 PLP2 [32]. Local protein mobility of NSP3-PLpro bound to Glycobismine F was evaluated by measuring residue time-averaged RMSF values on 50 ns trajectory data (Fig. S5G). The NSP3-PLpro residues bound to Glycobismine F showed less atomic fluctuation, suggesting that binding of Glycobismine F favors a more stable conformation.
A list of top ten compounds from NSP3-PLpro screening is summarized in Supplemental Table S1. Three promising compounds identified with high affinity free energy of − 9.3 kcal/mol include Albanol B, Mahuangnin A, and Rheidin B found in herbs Morus alba L., Murraya microphylla, and Rheum palmatum, respectively (Fig. 2C–E). Molecular docking of these ten compounds to the structure of SARS-CoV-2 PLpro (PDB 6XAA) showed comparable binding free energy, further supporting the highly conserved nature of this substrate-binding site of PLpro between SARS-CoV and SARS-CoV-2.
3.2 SARS-CoV-2 NSP5-Mpro
The SARS-CoV-2 main protease NSP5-Mpro, also referred to as 3CLPro, is a chymotrypsin-like cysteine protease. As the second key protease for viral polyprotein processing, it is responsible for releasing the rest of the replication complex subunit proteins (NSP 4–16). Therefore, NSP5-Mpro is an essential enzyme for SARS-CoV-2 viral function and is a critical drug target [33,34,35,36,37,38].
Recently published SARS-CoV-2 NSP5-Mpro crystal structures as apo protein (PDB 6M2Q) or in complex with the putative inhibitors (PDB 6LU7 and 6M2N) revealed that its overall structure consists of two β-barrel domains (domain I and II) and a helical bundle domain (domain III) [39]. The highly conserved substrate-binding site and the juxtaposed catalytic dyad with the signified residues Cys145 and His41 were identical to the previously reported SARS-CoV Mpro and MERS-CoV Mpro (Fig. 3A) [38, 40,41,42]. Surface analysis of SARS-CoV-2 NSP5-Mpro (PDB 6LU7) showed that the substrate-recognition pocket was located between the two β-barrel domains (domain I and II) responsible for anchoring the substrate peptide P2-Leu-P1-Gln↓(Ser, Ala, or Gly). NSP5-Mpro residues Asn142 and His163 form contacts with the substrate P1-Gln, and Met165 binds to the P2-Leu of the substrate [39]. Since this pocket conveys substrate recognition and specificity for SARS-CoV-2 NSP5-Mpro, it was used as a targeting site for this in silico screening.

The potential inhibitors of SARS-CoV-2 NSP5-Mpro identified by in silico screening. A The substrate-binding pocket located between the domain I and II was selected as the druggable pocket for NSP5-Mpro in silico screening. The catalytic dyad residues Cys145 and His41 were shown in red sticks. B The 2D and 3D structure of the identified compound DBGG as it was fitted in the binding pocket. The potential compound–protein interactions are depicted. C–E The 2D structures of three top-ranked compounds Ustilaginoidin A, bijaponicaxanthone, and Amentoflavone
Among the top ranked candidate compounds, 5,4-dihydroxyflavone-6-c-beta-glycosylrhamnoside-7-o-glycoside (DBGG) from the herb Glycyrrhiza yunnanensis (licorice root) was found to be deeply inserted into the substrate-binding pocket with affinity free energy of − 9.9 kcal/mol (Fig. 3B). This glycosyloxyflavone molecule forms multiple contacts with NSP5-Mpro including H-bonding with Asn142, His163, and the catalytic residue Cys145 at substrate P1 site as well as a hydrophobic interaction with Met165 at substrate P2 site. MD simulation was then performed on NSP5-Mpro bound to DBGG (Fig. S6) for 50 ns to study the potential interaction dynamics. RMSDs of NSP5-Mpro and DBGG revealed that the complex became stable after 25 ns of the MD run and finished with 0.18 nm for NSP5-Mpro and 0.43 nm for DBGG. RMSF of NSP5-Mpro bound to DBGG showed very small fluctuations throughout the structure, suggesting stable complex formation between DBGG and NSP5-Mpro. Analysis of the final computed NSP5-Mpro:DBGG complex confirmed the interaction of DBGG with NSP5-Mpro substrate-recognition pocket including residues Asn142 and Met165. As these contacts are critical in substrate binding and cleavage, this glycosyloxyflavone molecule is expected to affect NSP5-Mpro by steric hinderance of the active site.
A list of best-fitting compounds including DBGG for NSP5-Mpro was summarized in Supplementary Table S2. Among them, Ustilaginoidin A from Cldonia alpestris Rabht, Bijaponicaxanthone from Hypericum japonicum Thunb, and Amentoflavone from Ginkgo biloba (Fig. 3C-E) showed affinity free energy ranging from − 10.4 to − 10 kcal/mol and appeared to be better candidates than DBGG. However, closer examination of these three compounds revealed that they did not have contacts with key catalytic or substrate binding residues as DBGG. Amentoflavone has been previously reported to inhibit SARS-CoV Mpro [43, 44]. Further experimental validation of these candidate compounds will reveal their inhibitory activity against SARS-CoV-2 NSP5-Mpro.
3.3 SARS-CoV-2 NSP12-RdRp
SARS-CoV-2 is a positive strand RNA virus that relies on multi-subunit RNA synthesis machinery for genome replication and gene transcription. The core of its RNA replication complex is RdRp, produced from NSP12, one of the 16 cleavage products of the polyproteins pp1a and pp1ab. NSP12-RdRp is essential for SARS-CoV-2 replication and infection, therefore making it another desirable target for drug development [5].
The sequence alignment of SARS-CoV and SARS-CoV-2 NSP12-RdRp (protein ID: YP_009725307.1) revealed that these two proteins share 96% sequence identity and over 98% similarity (Fig. S3). The 3D structure of SAR-CoV NSP12-RdRp (PDB 6NUR chain A) was used as an initial template to build the SARS-CoV-2 NSP12-RdRp structure homology model by SWISS-MODEL used for in silico screening [45]. While the manuscript was in preparation, the EM structures of SARS-CoV-2 NSP12-RdRp in complex with co-factors were released (PDB 7BTF, 6M71, and 7BV2) [46]. Comparison of SARS-CoV-2 NSP12-RdRp with SARS-CoV NSP12-RdRp revealed RMSD of 0.4–0.6 Å over 931 NSP12-RdRp backbone Cα atoms with most differences located in the N-terminal NiRAN domain, whereas the core RNA polymerase domains (residues 366–920) remained highly conserved and almost identical. A large cup-shaped space could be found in the core RNA polymerase domains formed by the finger, palm, and thumb subdomains, containing the conserved Motif A-G for coordinating template and NTP for polymerization at the catalytic active site (Fig. 4A). A groove corresponding to NTP channel and the active site was selected as a targeting pocket for in silico screening, as is for the NSP12-RdRp inhibitor Remdisivir [46] (Fig. 4A).

The potential inhibitors of SARS-CoV-2 NSP12-RdRp identified by in silico screening. A The groove located at the NTP-binding site and the active site was selected as the druggable pocket for NSP12-RdRp in silico screening. The Thumb, Palm and finger subdomain were depicted. B The 2D and 3D structure of the identified Amentoflavone as it is fitted in the binding pocket. The potential compound–protein interactions are depicted. C–E. The 2D structures of another three top-ranked compounds include Astrochrysoside, Mulberrofuran M, and Ergocristine
Amentoflavone was identified to interact with NSP12-RdRp. It could fit into the selected pocket with affinity free energy of − 9.5 kcal/mol in the initial model built by SARS-CoV NSP12-RdRp (PDB 6NUR) and -9.7 kcal/mol using SARS-CoV-2 NSP12-RdRp (PDB 7BTF). As shown in Fig. 4B, Amentoflavone was inserted deep into the NTP pocket at the active site, forming H-bonds with the NTP coordinating residues Arg553 and Arg555 of Motif F, coordinating the active site residues Thr680, Ala685, Thr687, and Ala688 from Motif B, and also making π–cation interaction with R624 from Motif A. MD simulation was then performed on NSP12-RdRp bound to Amentoflavone (Fig. S7) for 50 ns to study the interaction dynamics. The complex became stable after 20 ns with the RMSD value for NSP12-RdRp stabilized at 0.25 nm and Amentoflavone at 0.2 nm. RMSF of NSP12-RdRp showed very small fluctuations around the Amentoflavone binding region (Residues 550—700), suggesting stable complex formation between Amentoflavone and NSP12-RdRp. MD analysis of Amentoflavone confirmed its interaction with the selected binding pocket of SARS-CoV-2 NSP12-RdRp, involving the residues Asp623 from Motif A, the residues Asp683, Ala685, Ala688 from Motif B and the residue Arg555 from Motif F (Fig. S7). As these contacts are critical for binding NTP and catalysis, Amentoflavone is expected to affect SARS-CoV2 NSP12-RdRp RNA polymerase activity by steric interference.
Among the top-ranked candidate compounds, Astrachrysoside from Astragalus chrysopterus, Mulbellofuran M from Morus alba L., and Ergocristine found in Claviceps purpurea (Fr.) Tul (Fig. 4C–E) appeared to fit in the targeted binding pocket better than Amentoflavone in the initial model with affinity free energy ranging from − 10.7 to − 9.6 kcal/mol. However, when validated with SARS-CoV-2 NSP12-RdRp (PDB 7BTF), these three compounds showed less binding affinity with SARS-CoV-2 NSP12-RdRp with free energy values ranging from − 8.2 to − 7.8 kcal/mol, suggesting the slight conformational difference of the active site between SARS-CoV and SARS-CoV-2 NSP12-RdRp. The information of these top-ranked candidate compounds for NSP12-RdRp and their binding affinity free energy values were summarized in Supplemental Table S3.
3.4 SARS-CoV2 NSP16-2OMT
SARS-CoV-2 NSP16-2OMT is one of the evolutionary conserved RNA processing enzymes encoded by SARS-CoV-2, responsible for viral mRNA capping. This enzyme catalyzes the last step of the viral RNA 5’-cap synthesis, converting Me7GpppN (cap 0 structure) to Me7GpppN-2O-Me (cap 1 structure) in S-adenosylmethione (SAM) dependent manner. SARS-CoV-2 NSP16-2OMT is essential for protecting viral mRNA from degradation and preventing it from being recognized by the host innate immune response [47]. Recent research has shown that the SARS virus with NSP16-2OMT mutations attenuated viral pathogenesis [48]. Thus, inhibition of SARS-CoV-2 NSP16-2OMT could intervene in viral RNA 2’-O-Methylation, promoting host immune defense and viral clearance.
The sequence alignment of SARS-CoV and SARS-CoV-2 NSP16-2OMT (protein ID: YP_009725311.1) revealed that these two proteins share 93% sequence identity and 97% similarity (Fig. S4). The 3D structure of SARS-CoV NSP16-2OMT (PDB 3R24 chain A) was used as an initial template to build the SARS-CoV-2 NSP16-2OMT structure homology model by SWISS-MODEL used for in silico screening [49]. The crystal structures of SARS-CoV-2 NSP16-2OMT in complex with NSP10 (PDB 6W4H, 6W61 and 6W75) were released while this manuscript was in preparation. Comparison of SARS-CoV-2 NSP16-2OMT (PDB 3R24 chain A) with SARS-CoV NSP16-2OMT (PDB 6W4H chain A) revealed the RMSD value of 0.24 Å over the backbone 294 Cα atoms of the structured region of the two NSP16s, whereas the SAM-binding and the active sites were mostly identical. A large “V” shaped space was found at the center of NSP16-2OMT. Within this space, the SAM binding cleft was formed by three loops depicted as SAM-L1, L2, and L3 on the right, the RNA-Cap groove on the left, and the catalytic residues Lys46, Asp130, Lys170, and Glu203 joining the SAM and RNA-Cap groove as a continuous channel [49] (Fig. 5A). Two runs of in silico screening were performed, the first run targeting the SAM-binding cleft and the second run focusing on the RNA-Cap groove. The top hit candidate compounds were then validated with the crystal structure of SARS-CoV-2 NSP16-2OMT (PDB 6W4H).

The potential inhibitors of SARS-CoV-2 NSP16-2OMT identified by in silico screening. A NSP16-2OMT structure was modeled based on SARS-CoV NSP16 (PDB 3R24 and 6W4H). Two pockets were selected for in silico screening. Pocket 1, SAM binding site (SAM-BS, left); Pocket 2, RNA-Cap binding groove (RNA-Cap-BS, right). SAM was shown in blue stick. The locations for SAM-Loop 1–3 and the catalytic residues Lys46, Asp130, Lys170 and Glu203 were indicated. B Ochnaflavone was identified from screening using SAM-binding site. The 2D and 3D structure of Ochnaflavone as it was fitted in the SAM binding pocket. The potential compound–protein interactions were depicted. C The 2D structures of another SAM-binding pocket compound Epicatechin-(2b- > 7,4b- > 6)-catechin. D. The 2D and 3D structure of Trichotomine as it was fitted in RNA-Cap binding pocket. E The 2D structures of another RNA-Cap binding pocket compound Mulberrofuran
In the virtual screening of SARS-CoV-2 NSP16-2OMT using the SAM-binding cleft, two structurally similar biflavonoid compounds Dihydroochnaflavone and Ochnaflavone were identified, with respective affinity free energy of − 10.8 kcal/mol and − 10.5 kcal/mol in the initial model, and − 10.4 kcal/mol and -10.1 kcal/mol in SARS-CoV-2 NSP16-2OMT (PDB 6W4H). As shown in Fig. 5B, Ochnaflavone fits into the SAM-binding groove, with one flavone ring forming H-bond and hydrophobic interactions with the residue Leu100 from SAM-L1 and the Met131 and Asp133 from SAM-L3, and the other flavone ring forming contacts with the catalytic residues Lys170 and Glu 203. MD simulation was performed on NSP16-2OMT bound to Ochnaflavone (Fig. S8) for 50 ns to explore the interaction dynamics. The NSP16-2OMT RMSD value was stabilized at 0.2 nm after 10 ns and the RMSD for the ligand Ochnaflavone was stabilized at 0.4 nm. RMSF of NSP16-2OMT showed atomic fluctuations (0.05–0.1 nm) around the binding region of Ochnaflavone (Residues 99–132 and 170–203), suggesting stable complex formation between NSP16-2OMT and Ochnaflavone. MD analysis of Ochnaflavone confirmed its interaction with the SAM-binding pocket of SARS-CoV-2 NSP16-2OMT including the residues Leu100, Met131 and Tyr132 (Fig. S8). As these residues are critical for NSP16-2OMT binding SAM, Ochnaflavone is expected to affect SARS-CoV2 NSP16-2OMT activity by steric interference.
As for the RNA-Cap binding pocket of SARS-CoV-2 NSP16-2OMT, Trichotomine from Clerodendron trichotomum was found to be the best scored compound with affinity free energy of − 10 kcal/mol. Trichotomine occupies the active site of NSP16-2OMT and extends toward the RNA-Cap entry site, forming contacts with the catalytic residues Lys170 and Glu203, as well as the key SAM/RNA-Cap binding residue Tyr132 (Fig. 5D). Importantly, as mutation of any of the catalytic residues or Tyr132 resulted in catalytically dead enzyme, both Ochnaflavone and Trichotomine are expected to block NSP16-2OMT enzymatic activities [49].
Additional candidate compounds for NSP16-2OMT include Epicatechin-(2b- > 7,4b- > 6)-catechin from Arachis hypogaea L. and Mulberrofuran K from Morus alba, which also have promising docking results (Fig. 5C, E, Supplemental Table S4). Epicatechin-(2b- > 7,4b- > 6)-catechin binds to the SAM binding site with affinity free energy of − 11.1 kcal/mol, whereas affinity free energy for Mulberrofuran K interaction with the RNA-Cap binding site appeared to be − 10.2 kcal/mol.
3.5 Pharmacokinetics prediction and evaluation
It is well known that an effective medicine not only needs to possess biological efficacy, but also good pharmacokinetic features, i.e. ability of a compound to reach therapeutic concentration in the organism. In this context, computer models were used to predict and evaluate the potential properties of a candidate compound in absorption, distribution, metabolism and excretion, referred as ADME, as an alternative approach to assist drug discovery and development. SwissADME was used to predict the physiochemical and pharmacokinetic properties of the identified candidate inhibitors for SARS-CoV-2 enzymes (Table 1, Supplementary Tables S5–S8). Among the top hits for SARS-CoV-2 enzymes, Glycobismine F, Amentoflavone, and Ochnaflavone presented high lipophilicity and low water solubility, while DBGG was predicted as a water-soluble molecule. All four compounds showed low skin permeability, low GI absorption, and no BBB permeation.
The pharmacokinetic relevant protein Permeability glycoprotein (P-gp) and cytochromes P450 (CYP) are the key drug metabolic enzymes. As inhibition of these enzymes is one major cause of drug toxicity and side effects due to low clearance, it is important to predict the compounds’ propensity of interaction with these pharmacokinetic relevant proteins. As shown in Table 1, Glycobismine F and Amentoflavone were not substrates or inhibitors of P-gp and CYP. DBGG was predicted to be the substrate of P-gp, whereas Ochnaflavone was shown to inhibit CYP isoform CYP2C9. Last, drug-likeness of the candidate compounds was evaluated by the probability of a molecule becoming an oral drug using the Lipinski filter (Pfizer) and Abbot bioavailability score [19]. Only Ochnaflavone met the Lipinski’s rules and showed properties of a good oral drug, while the other three were predicted as poor oral drug candidates.
4 Discussion
The economic and public health challenges that we are currently facing with the COVID-19 pandemic are unprecedented, resulting in unmet medical needs for effective therapeutics and prophylactic strategies to respond to the current and future outbreaks [50, 51]. Due to the fast evolving and heterologous nature of SARS coronavirus, inhibitors selectively targeting a single SARS-CoV-2 protein could be an untenable solution in the near term [52, 53]. On the other hand, in silico screening could provide a cost-effective method to identify bioactive molecules from different compound sources. A combination approach such as targeting multiple pathogenic pathways and viral proteins may represent a better strategy against the current virus outbreaks [54]. In this study, we performed in silico screening targeting four critical SARS-CoV-2 viral enzymes, NSP3-PLpro, NSP5-Mpro, NSP12-RdRp, and NSP16-2OMT, aiming to provide a pool of natural compounds with potential for attenuation of pathologic virulence of SARS-CoV-2 and prevention of COVID-19 infection.
To identify the potential inhibitors of these four SARS-CoV-2 viral enzymes, we targeted the primary substrate binding sites or active sites through in silico screening, as these sites are highly conserved and indispensable for the SARS-CoV-2 viral replication and function. An expanded library of 33,765 phytochemicals containing collections from TCM Database and bioactive compounds from herbs and medicinal plants was used to search for SARS-CoV-2 viral inhibitors [13]. Interestingly, several small molecules identified presented more than one potential viral targets (Supplemental Table S9). One such compound is Amentoflavone, a biflavonoid that is found particularly enriched in Ginkgo biloba and Hypericum perforatum. Amentoflavone was previously shown to be an inhibitor of cathepsin B, a lysosomal cysteine protease, and an antiviral agent against respiratory syncytial virus [55, 56]. Using virtual screening, we found that Amentoflavone could potentially inhibit both SARS-CoV-2 NSP-Mpro and NSP12-RdRp. In addition, the small molecule Albanol B was found to have affinity for both SARS-CoV-2 NSP3-PLpro and NSP12-RdRp. Although there was no direct evidence for its antiviral effect, Albanol B extracted from Mulberry (Morus alba L.) root bark was reported have an anti-inflammatory effect [57]. Thus, these small molecules could be promising antiviral compounds inhibiting multiple steps of the life cycle of SARS-CoV-2.
Furthermore, we summarized the potential active compounds identified in this study, which could be found in the herbs and medicinal plants from the herbal recipes recommended for COVID-19 prevention or treatment (Table 2 and Supplemental Table S10) [6, 8, 9, 58,59,60]. Notably, Mulberry root bark (Morus alba L., SANG BAI PI) and Licorice root (Glycyrrhiza uralensis, GAN CAO) appear to be the most popular herb ingredients used in herbal remedies against COVID-19, with potential active compounds affecting the activities of PLpro, Mpro, RdRp, and 2OMT, respectively. Licorice is one of the ancient remedy ingredients used for hepatitis and respiratory tract viral infection, currently under clinical trial as an antiviral therapy for COVID-19 infection [10,11,12]. Other active compounds identified from our study were found from popular medicinal herb and plant sources, including Ephedra (Herba ephedrae, MA HUANG), Agastache herb (Agastache rugosa, HUO XIANG), Honeysuckle Flower (Lonicera japonica, JING YIN HUA), Ginger (Zingiber officinale, JIANG), Milkvetch Root (Astragalus mongholicus Bunge, HUANG QI), Rhubarb root (Rheum officinale Baill. DA HUANG), Indigowoad Root (lsatis indigotica, BAN LAN GENG), and Fritillary bulb (Fritillaria thunbergii, BEI MU). Many of these ingredients have been historically used to treat respiratory viral infection [61, 62]. Similar remedies have shown benefits in combating SARS in 2003 [8, 9]. They may present novel antiviral effects against COVID-19 through inhibiting key enzymes of SARS-CoV-2. Optimization of these identified phytochemical compounds could lead to the discovery of more effective antiviral compounds and more powerful plant-based remedies targeting SARS-CoV-2.
References
Zhu N, Zhang D, Wang W et al (2020) A Novel Coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. https://doi.org/10.1056/NEJMoa2001017
Wu F, Zhao S, Yu B et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. https://doi.org/10.1038/s41586-020-2008-3
Zumla A, Hui DS, Perlman S (2015) Middle East respiratory syndrome. Lancet 386:995–1007. https://doi.org/10.1016/S0140-6736(15)60454-8
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol 92:418–423. https://doi.org/10.1002/jmv.25681
Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R (2020) COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J Adv Res 24:91–98. https://doi.org/10.1016/j.jare.2020.03.005
Runfeng L, Yunlong H, Jicheng H et al (2020) Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharmacol Res 156:104761. https://doi.org/10.1016/j.phrs.2020.104761
Wan S, Xiang Y, Fang W et al (2020) Clinical features and treatment of COVID-19 patients in northeast Chongqing. J Med Virol. https://doi.org/10.1002/jmv.25783
Yang Y, Islam MS, Wang J, Li Y, Chen X (2020) Traditional Chinese medicine in the treatment of patients infected with 2019-new coronavirus (SARS-CoV-2): a review and perspective. Int J Biol Sci 16:1708–1717. https://doi.org/10.7150/ijbs.45538
Ang L, Lee HW, Choi JY, Zhang J, Soo LM (2020) Herbal medicine and pattern identification for treating COVID-19: a rapid review of guidelines. Integr Med Res 9:100407. https://doi.org/10.1016/j.imr.2020.100407
Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW (2003) Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 361:2045–2046. https://doi.org/10.1016/s0140-6736(03)13615-x
Fiore C, Eisenhut M, Krausse R et al (2008) Antiviral effects of Glycyrrhiza species. Phytotherapy research : PTR 22:141–148. https://doi.org/10.1002/ptr.2295
Hoever G, Baltina L, Michaelis M et al (2005) Antiviral activity of glycyrrhizic acid derivatives against SARS-coronavirus. J Med Chem 48:1256–1259. https://doi.org/10.1021/jm0493008
Chen CY (2011) TCM Database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 6:e15939. https://doi.org/10.1371/journal.pone.0015939
Irwin JJ, Shoichet BK (2005) ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model 45:177–182. https://doi.org/10.1021/ci049714+
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminform 3:33. https://doi.org/10.1186/1758-2946-3-33
Guo R, Zhang Y, Li X, Song X, Li D, Zhao Y (2016) Discovery of ERBB3 inhibitors for non-small cell lung cancer (NSCLC) via virtual screening. J Mol Model 22:135. https://doi.org/10.1007/s00894-016-3007-z
Li X, Zhang Y, Chen H, Li H, Zhao Y (2017) Insights into the molecular basis of the acute contact toxicity of diverse organic chemicals in the honey bee. J Chem Inf Model 57:2948–2957. https://doi.org/10.1021/acs.jcim.7b00476
Weisel M, Proschak E, Schneider G (2007) PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem Cent J 1:7. https://doi.org/10.1186/1752-153X-1-7
Park H, Lee J, Lee S (2006) Critical assessment of the automated AutoDock as a new docking tool for virtual screening. Proteins 65:549–554. https://doi.org/10.1002/prot.21183
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Lill MA, Danielson ML (2011) Computer-aided drug design platform using PyMOL. J Comput Aided Mol Des 25:13–19. https://doi.org/10.1007/s10822-010-9395-8
Abraham MJ, Gready JE (2011) Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J Comput Chem 32:2031–2040. https://doi.org/10.1002/jcc.21773.
Sagui C, Darden TA (1999) Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu Rev Biophys Biomol Struct 28:155–179. https://doi.org/10.1146/annurev.biophys.28.1.155
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Shah S, Chaple D, Arora S, Yende S, Mehta C, Nayak U. Prospecting for Cressa cretica to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2. J Biomol Struct Dyn 2021:1–10. https://doi.org/10.1080/07391102.2021.1872419.
Bailey-Elkin BA, Knaap RC, Johnson GG et al (2014) Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem 289:34667–34682. https://doi.org/10.1074/jbc.M114.609644
Ratia K, Pegan S, Takayama J et al (2008) A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA 105:16119–16124. https://doi.org/10.1073/pnas.0805240105
Baez-Santos YM, St John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38. https://doi.org/10.1016/j.antiviral.2014.12.015
Bekes M, van der Heden van Noort GJ, Ekkebus R, Ovaa H, Huang TT, Lima CD (2016) Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Mol Cell 62:572–585. https://doi.org/10.1016/j.molcel.2016.04.016.
Ghosh AK, Xi K, Johnson ME, Baker SC, Mesecar AD (2007) Progress in Anti-SARS coronavirus chemistry, biology and chemotherapy. Annu Rep Med Chem 41:183–196. https://doi.org/10.1016/S0065-7743(06)41011-3
Baez-Santos YM, Barraza SJ, Wilson MW et al (2014) X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J Med Chem 57:2393–2412. https://doi.org/10.1021/jm401712t
Baez-Santos YM, Mielech AM, Deng X, Baker S, Mesecar AD (2014) Catalytic function and substrate specificity of the papain-like protease domain of nsp3 from the Middle East respiratory syndrome coronavirus. J Virol 88:12511–12527. https://doi.org/10.1128/JVI.01294-14
Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y (2021) Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res 52:38–47. https://doi.org/10.1016/j.arcmed.2020.09.013
Chen S, Chen L, Tan J, et al. (2005) Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. Biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J Biol Chem 280:164–173. https://doi.org/10.1074/jbc.M408211200.
Hsu MF, Kuo CJ, Chang KT et al (2005) Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem 280:31257–31266. https://doi.org/10.1074/jbc.M502577200
Huang C, Wei P, Fan K, Liu Y, Lai L (2004) 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 43:4568–4574. https://doi.org/10.1021/bi036022q
Shi J, Wei Z, Song J (2004) Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem 279:24765–24773. https://doi.org/10.1074/jbc.M311744200
Yang H, Yang M, Ding Y et al (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci U S A 100:13190–13195. https://doi.org/10.1073/pnas.1835675100
Jin Z, Du X, Xu Y et al (2020) Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature. https://doi.org/10.1038/s41586-020-2223-y
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767. https://doi.org/10.1126/science.1085658
Yang H, Xie W, Xue X et al (2005) Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3:e324. https://doi.org/10.1371/journal.pbio.0030324
Ho BL, Cheng SC, Shi L, Wang TY, Ho KI, Chou CY (2015) Critical assessment of the important residues involved in the dimerization and catalysis of MERS coronavirus main protease. PLoS ONE 10:e0144865. https://doi.org/10.1371/journal.pone.0144865
Ryu YB, Jeong HJ, Kim JH et al (2010) Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg Med Chem 18:7940–7947. https://doi.org/10.1016/j.bmc.2010.09.035
Schwarz S, Sauter D, Wang K et al (2014) Kaempferol derivatives as antiviral drugs against the 3a channel protein of coronavirus. Planta Med 80:177–182. https://doi.org/10.1055/s-0033-1360277
Kirchdoerfer RN, Ward AB (2019) Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 10:2342. https://doi.org/10.1038/s41467-019-10280-3
Gao Y, Yan L, Huang Y et al (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. https://doi.org/10.1126/science.abb7498
Chen Y, Su C, Ke M et al (2011) Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2’-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog 7:e1002294. https://doi.org/10.1371/journal.ppat.1002294
Menachery VD, Yount BL Jr, Josset L et al (2014) Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2’-o-methyltransferase activity. J Virol 88:4251–4264. https://doi.org/10.1128/JVI.03571-13
Decroly E, Debarnot C, Ferron F et al (2011) Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2’-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog 7:e1002059. https://doi.org/10.1371/journal.ppat.1002059
Abdullah, Faisal S, Aman K, Rahman AU (2020) Innate immune-mediated antiviral response to SARS-CoV-2 and convalescent sera a potential prophylactic and therapeutic agent to tackle COVID-19. Antib Ther 3:212–220. https://doi.org/10.1093/abt/tbaa019.
Jan H, Faisal S, Khan A et al (2020) COVID-19: review of epidemiology and potential treatments against 2019 novel coronavirus. Discoveries (Craiova) 8:e108. https://doi.org/10.15190/d.2020.5
Holmes EC, Rambaut A (2004) Viral evolution and the emergence of SARS coronavirus. Philos Trans R Soc Lond B Biol Sci 359:1059–1065. https://doi.org/10.1098/rstb.2004.1478
Giovanetti M, Angeletti S, Benvenuto D, Ciccozzi M (2020) A doubt of multiple introduction of SARS-CoV-2 in Italy: a preliminary overview. J Med Virol 92:1634–1636. https://doi.org/10.1002/jmv.25773
Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y (2021) SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: an in silico drug repurposing study. Eur J Pharmacol 896:173922. https://doi.org/10.1016/j.ejphar.2021.173922
Ma SC, But PP, Ooi VE et al (2001) Antiviral amentoflavone from Selaginella sinensis. Biol Pharm Bull 24:311–312. https://doi.org/10.1248/bpb.24.311
Pan X, Tan N, Zeng G, Zhang Y, Jia R (2005) Amentoflavone and its derivatives as novel natural inhibitors of human Cathepsin B. Bioorg Med Chem 13:5819–5825. https://doi.org/10.1016/j.bmc.2005.05.071
Wu YX, Kim YJ, Kwon TH, Tan CP, Son KH, Kim T. Anti-inflammatory effects of mulberry (Morus alba L.) root bark and its active compounds. Nat Prod Res 2018:1–4. https://doi.org/10.1080/14786419.2018.1527832.
Adhikari B, Marasini BP, Rayamajhee B et al (2021) Potential roles of medicinal plants for the treatment of viral diseases focusing on COVID-19: A review. Phytother Res 35:1298–1312. https://doi.org/10.1002/ptr.6893
Ang L, Lee HW, Kim A, Lee JA, Zhang J, Lee MS (2020) Herbal medicine for treatment of children diagnosed with COVID-19: a review of guidelines. Complement Ther Clin Pract 39:101174. https://doi.org/10.1016/j.ctcp.2020.101174
Luo H, Gao Y, Zou J et al (2020) Reflections on treatment of COVID-19 with traditional Chinese medicine. Chin Med 15:94. https://doi.org/10.1186/s13020-020-00375-1
Ding Y, Zeng L, Li R et al (2017) The Chinese prescription lianhuaqingwen capsule exerts anti-influenza activity through the inhibition of viral propagation and impacts immune function. BMC Complement Altern Med 17:130. https://doi.org/10.1186/s12906-017-1585-7
Zhou W, Zhang XY (2013) Research progress of Chinese herbal medicine Radix isatidis (banlangen). Am J Chin Med 41:743–764. https://doi.org/10.1142/S0192415X1350050X
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that there is no financial/personal interest or belief that could affect research objectivity and professional judgement concerning the validity of this research.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, Y., Tian, Y., Pan, C. et al. Target-Based In Silico Screening for Phytoactive Compounds Targeting SARS-CoV-2. Interdiscip Sci Comput Life Sci 14, 64–79 (2022). https://doi.org/10.1007/s12539-021-00461-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-021-00461-4