
Abstract
Background
The clinical manifestations of nonclassical 11beta-hydroxylase deficiency are very similar to those of non-classical 21-hydroxylase deficiency. For this study, we investigated the relationship between the clinical and molecular features of congenital adrenal hyperplasia caused by 11beta-hydroxylase deficiency and reviewed the related literature, which are expected to provide assistance for the clinical diagnosis and analysis of congenital adrenal hyperplasia.
Methods
Clinical data for 10 Chinese patients diagnosed with congenital adrenal hyperplasia in our hospital from 2018 to 2022 were retrospectively analyzed. We examined the effects of gene mutations on protease activity and constructed three-dimensional structure prediction models of proteins.
Results
We describe 10 patients with 11beta-hydroxylase gene mutations (n = 5, 46,XY; n = 5, 46,XX), with 10 novel mutations were reported. Female patients received treatment at an early stage, with an average age of 2.08 ± 1.66 years, whereas male patients received treatment significantly later, at an average age of 9.77 ± 3.62 years. The most common CYP11B1 pathogenic variant in the Chinese population was found to be c.1360C > T. All mutations lead to spatial conformational changes that affect protein stability.
Conclusions
Our study found that there was no significant correlation between each specific mutation and the severity of clinical manifestations. Different patients with the same gene pathogenic variant may have mild or severe clinical manifestations. The correlation between genotype and phenotype needs further study. Three-dimensional protein simulations may provide additional support for the physiopathological mechanism of genetic mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive diseases. The most common form of CAH originates from steroid 21-hydroxylase deficiency (21OHD), which accounts for about 90%–95% of all cases, followed by 11beta-hydroxylase deficiency (11βOHD) (OMIM 202010), which accounts for about 5%–8% of cases [1]. The incidence of 11β-OHD in the total population is approximately one in 100,000–200,000 [2, 3], and it is common in the Israeli population of Jewish origin in the Middle East and North Africa, affecting 1:5000–7000 live births [4]. 11β-OHD is caused by mutations in the cytochrome P450 family 11 subfamily B member 1 (CYP11B1) gene. CYP11B1 mutation leads to a reduction in the conversion of 11 deoxycorticosterone (DOC) to corticosterone and 11 deoxycorticosterol to cortisol, which leads to an increase in the secretion of adrenocorticotropic hormone (ACTH) and the accumulation of steroid precursors. Thus, 11β-OHD is characterized by increased serum concentrations of 11β-deoxycorticosterone, 17-hydroxyprogesterone (17-OHP) and androgens, resulting in hypertension with low renin levels, hypokalemia and genital ambiguity in affected individuals with a 46,XX karyotype. The milder nonclassical form is very similar to the nonclassical form of 21-OHD and is characterized by mild virilization, irregular menstruation, and male precocious puberty; hypertension does not occur frequently. According to the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk), a total of 155 CYP11B1 gene mutations were included, of which missense and nonsense mutations are among the most common types, and their effects on enzyme activity have been studied mainly through three-dimensional protein structure simulation and in vitro protein expression experiments.
Here, we report the clinical and molecular characterization of 10 Chinese patients with CAH due to 11β-OHD. Novel mutations and three previously reported but not functionally studied mutations were explored in vitro to monitor the residual enzyme activity and to analyze the correlation between the genotype and phenotype of the new mutations, providing help for clinical diagnosis and analysis of 11β-OHD. Three-dimensional protein simulations may provide additional support for the physiopathological mechanism of genetic mutations.
Methods
Subjects
Ten patients from unrelated Chinese families who had no history of consanguinity were enrolled. Some patients were first diagnosed with congenital adrenocortical insufficiency or CAH in other hospitals and then transferred to Ruijin Hospital for detailed examination. We gathered the documented clinical characteristics and blood biochemical examinations during each patient’s initial admission to the hospital (Ruijin Hospital or others) for CAH. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by the Ruijin Hospital Ethics Committee, Shanghai Jiao Tong University School of Medicine, and all the children and their guardians signed informed consent.
DNA sequence analysis of CYP11B1
Genomic DNA was extracted from peripheral blood using a DNA extraction kit (Qiagen, Hilden Germany) according to the manufacturer’s instructions. CYP11B1 gene (NM_000497.3) primers designed for polymerase chain reaction (PCR) were obtained from GenBank (Table 1). PCR products from primer pairs were resolved by 2% agarose gel electrophoresis and sequenced using an automated sequencer (ABI PRISM 3100 Genetic Analyzer; Applied Biosystems by Life Technologies, Carlsbad, CA, USA). Positive sequences were compared to the NCBI entry for CYP11B1 to confirm the success of site-directed mutagenesis.
Site-directed mutagenesis
A pcDNA3.1 expression vector construct with CYP11B1 complementary DNA (cDNA) as the insert (pcDNA3.1-CYP11B1 construct) was used as previously described. Single point mutations were introduced into CYP11B1 cDNA in the pcDNA3.1-CYP11B1-FLAG construct using PCR-based, site-directed mutagenesis and confirmed by first-generation sequencing.
Enzymatic activity assay
We studied the enzymatic activity of the 10 CYP11B1 mutants identified in this study, including some mutations that have been previously analyzed for function or have been reported but not functionally analyzed (Table 2). We resuscitated COS-7 cells were transfected using Lipofectamine 2000 reagent (Invitrogen, USA) with 1 µg of empty plasmid, wild-type CYP11B1 plasmid, or mutant CYP11B1 plasmid. After 48 hours of transfection, 1 mL of 11-deoxycortisol at 0.25, 0.5, or 1.0 μmol was added and incubated for 24 hours. The culture supernatant was collected, and the cortisol concentration was detected with an enzyme linked immunosorbent assay (ELISA) kit.
Western blots
COS-7 cells transfected with empty, wild-type CYP11B1, or mutant CYP11B1 plasmid were lysed in radio immunoprecipitation assay (RIPA) buffer (50 mmol/L Tris–Cl, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.1% sodium dodecyl sulfate (SDS), and 1% NP-40 pH 7.6) supplemented with a protease inhibitor cocktail (Roche, Switzerland). The lysates were denatured at 100 °C for 10 minutes in 1 × SDS- polyacrylamide gel electrophoresis (PAGE) loading buffer, subjected to SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, USA). The membrane was incubated with appropriate antibodies against FLAG (1:2000, 20,543–1-AP, Proteintech, USA) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000, 60,004–1-Ig, Proteintech, Chicago, USA). The secondary antibodies used were labeled with horse radish peroxidase (HRP), and the signals were visualized using a Tanon 5200 Imaging System (Tanon, Shanghai, China).
Construction of a three-dimensional structure prediction model of CYP11B1 protein
CYP11B1 missense mutations can affect the three-dimensional structure of the CYP11B1 enzyme, especially the three-dimensional conformation of the active site, resulting in decreased enzyme activity. Simulation of the three-dimensional structure of the protein at the above mutation sites may help to rationally explain and further confirm the results of the in vitro protein expression experiments.
The amino acid sequences of wild-type and mutant CYP11B1 were introduced into I-TASSER software to simulate the three-dimensional structure of the CYP11B1 variant protein with the X-ray structure of human CYP11B1 as a template [protein database (PDB accession number: 6M7X)] with visualization using PyMOL software.
Statistical methods
SPSS 22 was used for statistical analysis. Measurement data are expressed as the mean ± SD (standard deviation) or median. Height deviation was calculated using the Height Standard Deviation Score (SDS), which is calculated as (child height − normal child height)/normal child height SD. The same method was used for calculating weight.
Results
Clinical characteristics
The clinical characteristics of the patients are summarized in Table 2. The ten patients (five 46,XY and five 46,XX) were from 10 families. Age at diagnosis was delayed in the 46,XY patients (an average age of 9.77 ± 3.62 years) in comparison to 46,XX patients (an average age of 2.08 ± 1.66 years). All patients had hyperpigmentation. Among the five 46, XY male patients, premature adrenarche and peripheral precocious puberty were present at diagnosis in most cases, and macropenis was present in all male patients. Cases 3–5 involved hypokalemia and hypertension. As all female patients showed masculinization of external genitalia at birth, the age of treatment was earlier. Corticoid treatment consisted of hydrocortisone at diagnosis and during follow-up.
Hormone level characterization
The Hormonal level of the patients are summarized in Table 3. Seven patients were referred for androgen excess, and three were referred for hypertension and hypokalemia. Before treatment, adrenocorticotropic hormone (ACTH), cortisol, testosterone, aldosterone, renin, and electrolytes (including sodium and potassium) were detected in all patients, and dehydroepiandrosterone sulfate (DHEAS) and androstenedione (AD) were detected in six patients, and 17-OHP was detected in all but one patient. ACTH, 17-OHP, and the aldosterone/renin ratio were all elevated in all 10 patients without treatment; 5 patients had hypokalemia, and 2 patients had hypernatremia. An ACTH stimulation test was performed for 1 child (P9) without medication, the results showed that the baseline value of cortisol was low, The peak value of provocation was still low. The baseline value and peak value of 17-OHP and DHEAS were significantly higher than the normal range, and the baseline value and peak value of AD were also significantly increased.
Molecular analysis of the CYP11B1 gene
Through direct sequencing of PCR products, CYP11B1 gene mutations were detected in all 10 children; 3 cases involved homozygous mutations, and 7 were compound heterozygous mutations. There were four types of mutations in the 10 children, including four missense mutations (c.945C > A, c.950A > G, c.1355A > G, c.1360C > T), two nonsense mutations (c.421C > T, c.858C > A), five deletion mutations (c.363_365del, c.1391-1393delTGC, exon3-4 del, c.1150-1153delCGAG, c.1377-1378del), and one splice mutation (c.396-1G > A), for a total of 12 unique mutations. Excluding two point mutations and one nonsense mutation that have been described in other reports, these mutations have not been reported in the literature. The mutations included c.363_365del, c.858C > A, c.945C > A, c.950A > G, c.1391-1393delTGC, exon 3–4 del, c.1150-1153delCGAG, c.1377-1378del, and c.396-1G > A mutations. The mutation sites are mainly concentrated in exons 2, 5, 7, and 8. Consistent with previous reports, four patients (2, 5, 9, and 10) carried the c.1360C > T (p. R454C) mutation, which is found only in the Chinese population. The gene mutation results are shown in Table 2. A diagram of the CYP11B1 gene showing the location of mutations in 10 Chinese patients is provided in Fig. 1.

Diagram of the CYP11B1 gene showing the location of mutations in 10 Chinese patients
Amino acid conservation analysis of new missense mutations
Through multiple sequence alignment, we found that three new missense mutant amino acid sites of CYP11B1 to be highly conserved in mammals (Fig. 2), suggesting that mutations at these sites may have an important impact on protein function.

Comparison of the conservation of the missense mutation sites discovered this time among different species CYP11B1 cytochrome P450 family 11 subfamily B member 1
Identification of mutation sites for functional experiments and protein expression analysis
We reviewed relevant literature and summarized new mutations or CYP11B1 gene mutations that have been reported in the literature but have not been functionally studied, including c.400G > C (p. G134R), c.427C > T (p. R143W), c.456C > G (p. N152K), c.799G > T (p. G267S), c.896 T > C (p. L299P), c.945C > A (p. S315R), c.950A > T (p. D317V), c.1150C > G (p. R384G), c.1358G > A (p. R453Q), and c.1360C > T (p.R454C). Fig. 3a shows the locations of these CYP11B1 gene mutations. Expression levels of wild-type and mutant CYP11B1 were assessed by western blotting, and no obvious differences were observed (Fig. 3b).

Schematic localization and protein imprinting results of CYP11B1 mutation. a Diagram of these CYP11B1 genes showing the location of mutations; b Western blot results of CYP11B1 wild-type and mutant. WT wild-type, IB immunoblot, GAPDH glyceraldehyde-3-phosphate dehydrogenase
Residual enzyme activity results
The results of the enzymatic activity assay for the wild-type and CYP11B1 mutants proteins are shown in Fig. 4. The wild-type enzymatic activity was defined as 100%. The conversion percentages of 11-deoxycortisol (1 µmol/L) conversion were 11.5%, 12.3%, 12.3%, 6.7%, 9.4%, 10.4%, and 13.9%, respectively.

Enzymatic activity assay of wild-type and mutant CYP11B1. a Production of cortisol in the presence of various concentrations of 11-deoxycortisol (0.25, 0.5, 1.0 µmol) is shown as a measure of 11-hydroxylase activity in COS-7 cells transfected with the plasmid expressing the wild-type protein. b Compared with the wild-type enzyme, the 11-deoxycortisol (1 µmol/L) conversion percentages of the mutant enzymes were 11.5%, 12.3%, 12.3%, 6.7%, 9.4%, 10.4%, and 13.9% for c.400G > C (p. G134R), c.427C > T (p. R143W), c.456C > G (p. N152K), c.896 T > C (p. L299P), c.950A > T (p. D317V), c.1150C > G (p. R384G), c.1358G > A (p. R453Q), respectively
CYP11B1 mutation function prediction and three-dimensional protein structure prediction model
The amino acid conservation analysis of CYP11B1 in 7 species was carried out through the Uniprot database, and it was found that S315 does not exhibit high conservation; N152 is a variable amino acid; D317 and R454 are highly conserved residues. Then we used PolyPhen-2, SIFT and CADD software to predict whether these four different missense mutation would be harmful to the protein function. The results showed that S315R, D317V and R454C are damaging, while N152K is benign (Table 4).
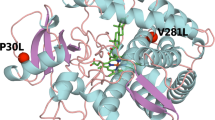
Figure 5 is a three-dimensional protein structure prediction model for CYP11B1. The protein model prediction showed that the p.S315R mutant did not form hydrogen bonds with T312 but increased the hydrogen bonds with S200 (Fig. 5a); the p.N152K mutant was the same as the wild type, and both only formed hydrogen bonds with R156 (Fig. 5b); the p.D317V increases hydrogen bonding with F321, which may enhance the local interaction strength and reduce the flexibility of the domain (Fig. 5c); and p.R454C increases hydrogen bonding with L451, which may enhance local interaction strength and reduce domain flexibility (Fig. 5d).

CYP11B1 three-dimensional protein structure prediction model. White: wild-type; cyan: mutant; red: mutation site. Left: wild type, right: mutant
Discussion
In this study, we describe the clinical and molecular characterization of 11β-hydroxylase deficiency and the genotype–phenotype correlation of 11β-OHD in 10 Chinese patients from 10 unrelated families. Five female patients were affected by excessive fetal adrenal androgens in the uterus, resulting in ambiguous external genitalia and varying degrees of masculinization at birth [5]. Therefore, the female patients were treated at an early age, at an average age of 2.08 ± 1.66 years old. The age of the 46,XY patients at the time of diagnosis ranged from 4.25 to 12 years, with the difference between bone age and chronological age being 3.69 ± 2.82 years. The age of diagnosis and treatment of female patients was significantly earlier than that of male patients. All 10 patients displayed different degrees of skin pigmentation. Four patients were initially misdiagnosed with 21-OHD. Due to the increase in DOC, some patients develop hypertension with low aldosterone and low renin with hypokalemia. Therefore, the levels of DOC and deoxycortisol are important diagnostic criteria for 11β-OHD. Unfortunately, they are not routinely detected in China. In our clinical practice, hypertension is the main clue to differentiate 11β-OHD from 21-OHD. However, most patients do not have manifestations of hypertension and hypokalemia at birth, and some patients do not have hypertension [6, 7]. Therefore, it is generally detected later in childhood or even in adolescence. In our study, three male patients presented hypertension and hypokalemia due to delayed diagnosis of 11β-OHD and delayed treatment. The female patients were diagnosed at an early age and were treated in a timely manner without hypertension or hypokalemia. Complications such as cardiomyopathy, retinal vein occlusion, and even blindness from long-lasting, uncontrolled hypertension have been reported in patients with 11β-OHD [8]. Early diagnosis and treatment are key points to prevent the complications of hypertension. The lack of negative feedback of glucocorticoids leads to increased secretion of ACTH, such that adrenal crisis rarely occurs, but patients generally have hyperpigmentation. Therefore, molecular analysis of the CYP11B1 gene is particularly important to confirm a suspected clinical diagnosis of 11β-OHD.
11β-OHD is caused by mutations in CYP11B1 located on chromosome 8q21-22. The CYP11B1 gene consists of nine exons and encodes a 503 amino acid protein. To date, the HGMD has reported 98 missense and nonsense mutations. These mutations are concentrated in exons 2 and 6–8, with a small number in exons 1 and 9. In this study, we detected 13 mutations, including three point mutations and one nonsense mutation that have been described in other reports [9], of which nine mutations are novel (Y286X, S315R, D317V, Q121del, L464del, exon 3–4 deletion, R384Wfs × 45, E459fs, c.396-1G > A). The mutation sites are mainly concentrated in exons 2, 5, 7, and 8. Consistent with previous reports, four patients (P2, P5, P9, P10) carried the R454C pathogenic variation, which is found only in the Chinese population [10]. The nine novel mutations included two missense mutations (S315R and D317V), one nonsense mutation (Y286X), five deletion frameshifts (Q121del, L464del, exon 3–4 deletions, R384Wfs × 45 and E459fs) and one splice mutation (c.396-1G > A). Nonsense mutations, deletion frameshift mutations and splicing mutations all indicate loss of enzymatic activity. Therefore, newly discovered and previously reported missense mutations that have not been tested for enzymatic activity were selected for enzymatic activity function detection, and the encoded enzymatic protein after gene mutation was analyzed. For the D317V mutation detected in patient 4, enzyme activity was 9.4% (< 10%); the patient showed clinically accelerated growth with peripheral precocious puberty, which was misdiagnosed as 21-OHD in other hospitals, and developed hypertension and hypokalemia several years later. Therefore, the disease was considered to be classic 11β-OHD. For patient 8 (46, XX), N152K mutant enzyme activity was 12.3%, this patient had obvious masculinization of external genitalia at birth with renin in infancy of 0.1 ng/mL/h, and serum potassium of 3.67 mmol/L. Therefore, the case was also considered classical 11β-OHD. In 2010, Parajes et al. [11] detected M88I and P159L mutations in patients with the nonclassical form; these mutations decreased the enzyme activity to 40% and 25% of the wild-type level, respectively. While studies have shown that CYP11B1 mutations that cause classic 11β-OHD usually reduce enzyme activity to less than 5% or result in complete loss of activity, for each specific mutation, there is no significant correlation with the severity of the clinical manifestations for each specific mutation. Indeed, patients with the same gene pathogenic variant may have mild or severe hypertension, and the manifestations of androgen excess vary in severity. Thus, the correlation between genotype and phenotype needs to be further studied.
Through the three-dimensional protein structure model, we attempted to infer the influence of the mutation site on the three-dimensional structure of the protein conformation. The helix structure is the heme-binding region and is highly conserved. The cysteine sulfhydryl group at position 450 binds with a heme iron atom to form one of the enzyme active sites. Therefore, the amino acid changes around C450 can affect binding of heme and cause the loss of enzyme activity. For example, Y423X, Q426X, P427H, V441G, G444D, G446V, G446S, R448C, R448H, R448P, R453Q, R454C, R461P and other mutations can cause a loss of enzyme activity [10, 12], of which R454C has been reported only in the Chinese population. The R454 residue is highly conserved; protein model predictions show increased hydrogen bonding with L451 for the p.R454C mutant, which may enhance local interaction strength and reduce domain flexibility.
As part of the substrate binding pocket, the I-helix contains many hydrophobic amino acids and potential enzyme active sites and is involved in the recognition of and binding to substrates. When its conformation changes, the enzyme activity of CYP11B1 may be impaired. For example, L299P, A368D, P94L, A331V, E371G and other mutations can reduce or even eliminate the activity of the enzyme [13,14,15]. The G-helix, the I-helix and the B-C loop between them are important channels for the substrate to enter the active site the G-helix is necessary for the initial recognition of the substrate, and the change in the hydrophobic environment around it can hinder the binding of the substrate to the enzyme. For example, W116C, W116G, V129M, V148G, A259D, T318M, T318R, T318P, R384Q, R384G and other mutations can reduce or even cause loss of enzyme activity [15]. The c.945C > A mutation was detected in patient 1, resulting in the amino acid change S315R (Ser315Arg), which is a new missense mutation. The amino acid conservation analysis of CYP11B1 in seven species was carried out through the UniProt database, and S315 is not highly conserved; PolyPhen-2 (score 1.000) software predicts the p. S315R variant to be deleterious, but with SIFT (score 0.073) and CADD (score 17.80) software, the deleterious threshold was not reached. The protein model prediction showed that the p.S315R mutant does not form hydrogen bonds with T312 but shows increased the hydrogen bonding with S200. The c.456C > G mutation was detected, leading to the amino acid N152K (Asn315Lys), was detected in patient 8. This missense mutation has been reported, but no functional experiments have been performed.Amino acid conservation analysis of CYP11B1 in seven species was performed through the UniProt database. N152 is a variable amino acid; PolyPhen-2 (score 0.000), SIFT (score 1.000), and CADD (score 0.024) predict that the p. N152K variant to be benign. The protein model prediction showed that the p. N152K mutant folds in the same manner as the wild-type protein, and in both cases, the residue at this position forms hydrogen bonds only with R156. This patient is 46,XX, with clinical manifestations of clitoral hypertrophy, visible vaginal opening, urethral opening, Prader stage II, and Tanner stage B1PH1, but no manifestations of hypertension. Therefore, this current missense mutation of CYP11B1 can lead to changes in the three-dimensional conformation of the CYP11B1 enzyme, leading to different degrees of changes in enzyme activity.
In conclusion, c.1360C > T is the most common CYP11B1 pathogenic variant in the Chinese population. Enzymatic activity assays combined with clinical characteristics showed a good clinical phenotype-genotype correlation in this study. Three-dimensional protein simulations may provide additional support for the physiopathological mechanism of genetic mutations. Further research is still needed in the future.
Data availability
All data generated or analyzed during this study are included in this published article.
Change history
19 January 2024
A Correction to this paper has been published: https://doi.org/10.1007/s12519-023-00784-w
References
Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency A study of 25 patients. J Clin Endocrinol Metab. 1983;56:222–9.
White PC, Obeid J, Agarwal AK, Tannin GM, Nikkila H. Genetic analysis of 11 beta-hydroxysteroid dehydrogenase. Steroids. 1994;59:111–5.
Khattab A, Haider S, Kumar A, Dhawan S, Alam D, Romero R, et al. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. Proc Natl Acad Sci USA. 2017;114:E1933–40.
Khemiri M, Ridane H, Bou YO, Matoussi N, Khaldi F. 11 beta hydroxylase deficiency: a clinical study of seven cases. Tunis Med. 2006;84:106–13.
Reisch N, Hogler W, Parajes S, Rose IT, Dhir V, Gotzinger J, et al. A diagnosis not to be missed: nonclassic steroid 11beta-hydroxylase deficiency presenting with premature adrenarche and hirsutism. J Clin Endocrinol Metab. 2013;98:E1620–5.
Rosler A, Leiberman E, Cohen T. High frequency of congenital adrenal hyperplasia (classic 11 beta-hydroxylase deficiency) among Jews from Morocco. Am J Med Genet. 1992;42:827–34.
Burren CP, Montalto J, Yong AB, Batch JA. CYP11 beta 1 (11-beta-hydroxylase) deficiency in congenital adrenal hyperplasia. J Paediatr Child Health. 1996;32:433–8.
Chabre O, Portrat-Doyen S, Chaffanjon P, Vivier J, Liakos P, Labat-Moleur F, et al. Bilateral laparoscopic adrenalectomy for congenital adrenal hyperplasia with severe hypertension, resulting from two novel mutations in splice donor sites of CYP11B1. J Clin Endocrinol Metab. 2000;85:4060–8.
Ye ZQ, Zhang MN, Zhang HJ, Jiang JJ, Li XY, Zhang KQ. A novel missense mutation, GGC(Arg454)–> TGC(Cys), of CYP11B1 gene identified in a Chinese family with steroid 11beta-hydroxylase deficiency. Chin Med J (Engl). 2010;123:1264–8.
Wu C, Zhou Q, Wan L, Ni L, Zheng C, Qian Y, et al. Novel homozygous pR454C mutation in the CYP11B1 gene leads to 11beta-hydroxylase deficiency in a Chinese patient. Fertil Steril. 2011;95:1122.
Parajes S, Loidi L, Reisch N, Dhir V, Rose IT, Hampel R, et al. Functional consequences of seven novel mutations in the CYP11B1 gene: four mutations associated with nonclassic and three mutations causing classic 11beta-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:779–88.
Krone N, Grotzinger J, Holterhus PM, Sippell WG, Schwarz HP, Riepe FG. Congenital adrenal hyperplasia due to 11-hydroxylase deficiency-insights from two novel CYP11B1 mutations (pM92X, pR453Q). Horm Res. 2009;72:281–6.
Krone N, Grischuk Y, Muller M, Volk RE, Grotzinger J, Holterhus PM, et al. Analyzing the functional and structural consequences of two point mutations (P94L and A368D) in the CYP11B1 gene causing congenital adrenal hyperplasia resulting from 11-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:2682–8.
Riedl S, Nguyen HH, Clausmeyer S, Schulze E, Waldhauser F, Bernhardt R. A homozygous L299P mutation in the CYP11B1 gene leads to complete virilization in 46, XX individuals with 11-beta-hydroxylase deficiency. Horm Res. 2008;70:145–9.
Zhao LQ, Han S, Tian HM. Progress in molecular-genetic studies on congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. World J Pediatr. 2008;4:85–90.
Acknowledgements
We gratefully acknowledge the contribution of children and parents, research employees in this study
Funding
None.
Author information
Authors and Affiliations
Contributions
LWL: conceptualization, funding acquisition, investigation, methodology, visualization, writing–original draft. MXY: conceptualization, investigation, methodology, visualization. ZJ: conceptualization, investigation, methodology, visualization. WJQ: data curation, formal analysis, resources. ZTT: data curation, formal analysis. YL Xiao Y, DZY, WW, SSY: project administration, validation. LCY, HRG: conceptualization, supervision. NG: conceptualization, writing–review and editing. ZLD: conceptualization, writing–original draft, writing–review and editing. LWL, MXY and ZJ contributed equally to this paper.
Corresponding authors
Ethics declarations
Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
Ethical approval
The Medical Ethics Committee of Ruijin Hospital approved the study, and the approval form is attached in the supplementary materials.. Written consent for publication of the case details together with imaging or videos have been obtained from the children and their guardians.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article contained an error in Fig. 2. Figure 2 in the original version shows a comparison of missense mutation sites found between different steroidogenic acute regulatory (STAR) proteins from the indicated species.
The authors correct Fig. 2 as a comparison of the conservation of missense mutation sites discovered between different species of cytochrome P450 family 11 subfamily B member 1 (CYP11B1) proteins.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, WL., Ma, XY., Zhang, J. et al. Clinical and molecular characterization of 10 Chinese children with congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. World J Pediatr 20, 422–433 (2024). https://doi.org/10.1007/s12519-023-00739-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-023-00739-1