
Abstract
Background
The PACS gene family has been demonstrated to be related to intracellular vesicular trafficking. The phenotypic manifestations caused by the pathogenic variants of PACS include epilepsy, intellectual disability/developmental delay, and malformations, such as facial abnormalities.
Methods
We identified seven new cases with pathogenic or likely pathogenic PACS variants using next-generation sequencing. Detailed information obtained from these patients was analyzed along with that obtained from previously reported patients.
Results
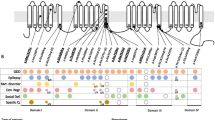
With the inclusion of the newly diagnosed cases in this study, 103 cases with PACS gene family-related neurological diseases were reported, of which 43 were PACS2-related cases and the remaining were PACS1-related cases. Most patients had seizures, which have been reported to be effectively controlled by several types of anti-seizure medications (ASMs). The most efficacious and frequently prescribed ASMs included sodium valproate (43.3%, 13/30), oxcarbazepine/carbamazepine (26.7%, 8/30), and levetiracetam (20%, 6/30). Almost all patients had intellectual disability/developmental delay. The most common pathogenic missense variants were PACS1 p. Arg203Trp and PACS2 p.Glu209Lys. In addition, we report a patient carrying a likely pathogenic copy number variation (CNV) (de novo heterozygous deletion of chr14:105821380-106107443, 286 kilobase, destroyed part of the furin-binding region domain and the protein structure after it) with more severe and refractory late-onset epilepsy.
Conclusions
The clinical phenotypes of the different PACS heterozygous missense variants were similar. The pathogenic variant sites of PACS1 and PACS2 were quite limited but located in different regions. A CNV destroying part of the PACS2 gene might also be pathogenic. These findings may provide an important clue for further functional studies on the pathogenic mechanism of neurological disorders related to the PACS gene family.
Video Abstract (MP4 65767 kb)


Similar content being viewed by others


Data availability
Most data generated or analyzed during this study are included in this published article. Other data are available from the corresponding author on reasonable request.
References
Li C, Li L, Yang M, Zeng L, Sun L. PACS-2: a key regulator of mitochondria-associated membranes (MAMs). Pharmacol Res. 2020;160:105080.
Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94:205–16.
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24:717–29.
Olson HE, Jean-Marçais N, Yang E, Heron D, Tatton-Brown K, van der Zwaag PA, et al. A recurrent de novo PACS2 heterozygous missense variant causes neonatal-onset developmental epileptic encephalopathy, facial dysmorphism, and cerebellar dysgenesis. Am J Hum Genet. 2018;102:995–1007.
Dentici ML, Barresi S, Niceta M, Ciolfi A, Trivisano M, Bartuli A, et al. Expanding the clinical spectrum associated with PACS2 mutations. Clin Genet. 2019;95:525–31.
Mizuno T, Miyata R, Hojo A, Tamura Y, Nakashima M, Mizuguchi T, et al. Clinical variations of epileptic syndrome associated with PACS2 variant. Brain Dev. 2021;43:343–7.
Olson HE, Jean-Marçais N, Yang E, Heron D, Tatton-Brown K, van der Zwaag PA, et al., A recurrent de novo PACS2 heterozygous missense variant causes neonatal-onset developmental epileptic encephalopathy, facial dysmorphism, and cerebellar dysgenesis. Am J Hum Genet. 2018;103:631.
Terrone G, Marchese F, Vari MS, Severino M, Madia F, Amadori E, et al. A further contribution to the delineation of epileptic phenotype in PACS2-related syndrome. Seizure. 2020;79:53–5.
Zhu T, Gong X, Bei F, Ma L, Chen Y, Zhang Y, et al. Application of next-generation sequencing for genetic diagnosis in neonatal intensive care units: results of a multicenter study in China. Front Genet. 2020;11:565078.
Wu MJ, Hu CH, Ma JH, Hu JS, Liu ZS, Sun D. Early infantile epileptic encephalopathy caused by PACS2 gene variation: three cases report and literature review. Zhonghua Er Ke Za Zhi. 2021;59:594–9 (in Chinese).
Wang T, Hoekzema K, Vecchio D, Wu H, Sulovari A, Coe BP, et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat Commun. 2020;11:4932.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104:1182–201.
Jiang T, Gao J, Jiang L, Xu L, Zhao C, Su X, et al. Application of trio-whole exome sequencing in genetic diagnosis and therapy in Chinese children with epilepsy. Front Mol Neurosci. 2021;14:699574.
Bruel AL, Nambot S, Quéré V, Vitobello A, Thevenon J, Assoum M, et al. Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. Eur J Hum Genet. 2019;27:1519–31.
Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, Swinkels ME, Gilissen C, Willemsen MA, et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012;91:1122–7.
Seto MT, Bertoli-Avella AM, Cheung KW, Chan KY, Yeung KS, Fung JL, et al., Prenatal and postnatal diagnosis of Schuurs-Hoeijmakers syndrome: case series and review of the literature. Am J Med Genet A. 2021;185:384–9.
Tenorio-Castaño J, Morte B, Nevado J, Martinez-Glez V, Santos-Simarro F, García-Miñaúr S, et al. Schuurs-Hoeijmakers syndrome (PACS1 neurodevelopmental disorder): seven novel patients and a review. Genes (Basel). 2021;12:738.
Martinez-Monseny A, Bolasell M, Arjona C, Martorell L, Yubero D, Arsmtrong J, et al. Mutation of PACS1: the milder end of the spectrum. Clin Dysmorphol. 2018;27:148–50.
Hoshino Y, Enokizono T, Imagawa K, Tanaka R, Suzuki H, Fukushima H, et al. Schuurs-Hoeijmakers syndrome in two patients from Japan. Am J Med Genet A. 2019;179:341–3.
Thomas G, Aslan JE, Thomas L, Shinde P, Shinde U, Simmen T. Caught in the act-protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J Cell Sci. 2017;130:1865–76.
Miyake N, Ozasa S, Mabe H, Kimura S, Shiina M, Imagawa E, et al. A novel missense mutation affecting the same amino acid as the recurrent PACS1 mutation in Schuurs-Hoeijmakers syndrome. Clin Genet. 2018;93:929–30.
Holder JL Jr, Lotze TE, Bacino C, Cheung SW. A child with an inherited 0.31 Mb microdeletion of chromosome 14q32.33: further delineation of a critical region for the 14q32 deletion syndrome. Am J Med Genet A. 2012;158A:1962–6.
Acknowledgements
The authors thank the patients mentioned in the article and their families for the clinical information they provided and their contribution to scientific research.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2020YFA0804000), the National Natural Science Foundation of China (Nos. 81971211, 12026606, and 81601131), Key Project of Clinical Medicine Research of National Clinical Research Center for Child Health and Disorders, Children’s Hospital of Chongqing Medical University (No. NCRCCHD-2021-KP-02), Beijing Natural Science Foundation (No. 7212109), the Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (No. BZ0317), the Capital Health Research and Development of Special Fund (No. 2020–1-4071), National High Level Hospital Clinical Research Funding (Scientific Research Seed Fund of Peking University First Hospital) (No. 2022SF29), and the Fundamental Research Funds for the Central Universities (Nos. BMU2017JI002, BMU2018XY006, and PKU2017LCX06). The funding agencies had no role in the study design, the experiments, analysis, or interpretation of data, the writing of the report, or the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
ZH and GK contributed equally to this manuscript and they were co-first authors. ZH contributed to study design, collection, evaluation and interpretation of the data, and drafting and writing of the manuscript. WS, ZYH, YZX, and WY contributed to collection, evaluation and interpretation of the data. GK and JYW contributed to study design, collection, evaluation and interpretation of the data, and revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethical approval
This study was approved by the Ethical Committee of Peking University First Hospital. The individuals’ parents in this manuscript had given written informed consent to publish these cases details. All data were analyzed anonymously.
Conflict of interest
Author Yu-Wu Jiang is a member of Editorial Board for World Journal of Pediatrics. The paper was handled by the other Editor and has undergone rigorous peer review process. Author Yu-Wu Jiang was not involved in the journal's review of, or decisions related to this manuscript. All authors declared no financial or non-financial conflict of interests related to this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, H., Gao, K., Wang, S. et al. PACS gene family-related neurological diseases: limited genotypes and diverse phenotypes. World J Pediatr 20, 82–91 (2024). https://doi.org/10.1007/s12519-022-00652-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-022-00652-z