
Abstract
Background
Enzyme replacement therapy (ERT) has been increasingly used as an interim treatment in severe mucopolysaccharidosis type I (MPSI)/Hurler patients prior to hematopoietic stem cell transplantation (HSCT).
Methods
We present the outcome of a patient with MPSI/Hurler after 14 months of ERT prior to HSCT.
Results
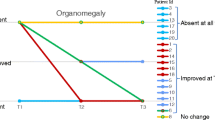
Urinary glucosaminoglycan excretion decreased by 70% after one month of ERT. Liver volume decreased by 14% of baseline after 12 months of ERT. Pre-existing thoracolumbar kyphosis progressed to thoracolumbar dislocation with complete displacement of facets after 12 months of ERT. New development of mitral valve thickening was found by echocardiography and mild hearing loss progressed to severe sensorineural hearing loss after 13 months of ERT.
Conclusions
ERT over a period of 14 months did not prevent progression of organ manifestations in our patient. Patients should be monitored every 6 months for cardiac, skeletal and audiological involvement on ERT.
Similar content being viewed by others

References
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic & molecular bases of inherited disease. New York: McGraw-Hill, 2001: 3421–3452.
Peters C, Balthazor M, Shapiro EG, King RJ, Kollman C, Hegland JD, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996;87:4894–4902.
Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004;144:581–588.
Grewal SS, Wynn R, Abdenur JE, Burton BK, Gharib M, Haase C, et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet Med 2005;7:143–146.
Scott HS, Litjens T, Nelson PV, Brooks DA, Hopwood JJ, Morris CP. alpha-L-iduronidase mutations (Q70X and P533R) associate with a severe Hurler phenotype. Hum Mutat 1992;1:333–339.
Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, et al. A follow-up study of MPSI patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 2007;90:171–180.
Braunlin EA, Berry JM, Whitley CB. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am J Cardiol 2006;98:416–418.
Tokic V, Barisic I, Huzjak N, Petkovic G, Fumic K, Paschke E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur J Pediatr 2007;166:727–732.
Thomas JA, Jacobs S, Kierstein J, Van Hove J. Outcome after three years of laronidase enzyme replacement therapy in a patient with Hurler syndrome. J Inherit Metab Dis 2006;29:762.
Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001;344:182–188.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mercimek-Mahmutoglu, S., Reilly, C., Human, D. et al. Progression of organ manifestations upon enzyme replacement therapy in a patient with mucopolysaccharidosis type I/Hurler. World J Pediatr 5, 319–321 (2009). https://doi.org/10.1007/s12519-009-0062-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-009-0062-x