
Abstract
Introduction
Once-weekly (OW) subcutaneous (s.c.) semaglutide is an injectable glucagon-like peptide-1 (GLP-1) analogue approved for the treatment of type 2 diabetes. This trial was designed to assess the pharmacokinetics, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects.
Methods
In this single-centre, randomised, double-blind, placebo-controlled trial, 36 healthy subjects were randomised to OW s.c. semaglutide 0.5 mg (n = 12), 1.0 mg (n = 12), or placebo (n = 12). Treatment (semaglutide or placebo) was blinded for the subjects, investigators and sponsor. The primary endpoint was steady-state semaglutide exposure, defined as the area under the curve over a dosing interval at steady state (AUC0–168 h,SS).
Results
In total, 34 subjects completed the trial. The steady-state exposure of semaglutide was higher for subjects treated with 1.0 mg semaglutide (AUC0–168 h,ss: 7961 nmol h/l and Cmax,ss: 55.9 nmol/l) compared to 0.5 mg semaglutide (AUC0–168 h,ss: 4000 nmol h/l and Cmax,ss: 28.8 nmol/l). The total exposure of semaglutide increased in a dose-proportional manner in healthy Chinese subjects; the treatment ratio (1.0 mg/0.5 mg) [95% confidence interval] for AUC0–168 h,SS was 1.99 [1.78; 2.23]. Treatment with OW s.c. semaglutide was well tolerated in healthy Chinese subjects. As expected for the GLP-1 receptor agonist class, the most common adverse events were gastrointestinal, and no new safety signals were identified.
Conclusion
The pharmacokinetics, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects were consistent with previous clinical pharmacology trials of OW s.c. semaglutide in other populations. The results suggest that no dose adjustment is necessary for semaglutide in Chinese patients with T2D.
Trial Registration
ClinicalTrials.gov, identifier NCT03288740.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Despite the availability of numerous glucose-lowering drugs, < 20% of Chinese patients with type 2 diabetes (T2D) achieve the recommended glycaemic control target (HbA1c < 7%) and there is an unmet need for an effective, convenient and easy-to-use treatment option for patients with T2D. |
Semaglutide (Ozempic®; Novo Nordisk A/S, Denmark) is a glucagon-like peptide-1 (GLP-1) analogue, approved in numerous countries for the once weekly (OW) subcutaneous (s.c.) treatment of T2D; the large SUSTAIN programme demonstrated that OW s.c. semaglutide provided good glycaemic control versus a wide range of comparators, in addition to providing additional clinical benefits such as weight loss, cardiovascular risk reduction and lowering of systolic blood pressure, with a low risk of hypoglycaemia. |
This trial was designed to assess the pharmacokinetics, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects. |
What was learned from the study? |
The pharmacokinetics, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects were consistent with previous clinical pharmacology trials of OW s.c. semaglutide in other populations. |
The results suggest that no dose adjustment is necessary for semaglutide in Chinese patients with T2D. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.13123361.
Introduction
Type 2 diabetes (T2D) is a progressive, chronic, metabolic disease characterised by persistent hyperglycaemia which, if left untreated, is associated with increased risk of long-term microvascular and macrovascular complications [1]. T2D is associated with many comorbidities, including obesity and cardiovascular disease [2, 3]. In addition, diabetes is an independent risk factor for cardiovascular disease and, alongside smoking, obesity, dyslipidaemia and hypertension, is one of the most important risk factors for heart disease and stroke worldwide [3, 4]. Despite the severity of the disease, a large proportion of patients with T2D do not achieve glycaemic targets [5,6,7].
China has the largest population of patients with T2D in a single country [1]. Among adults in China in 2017, the estimated overall prevalence of diabetes was 10.9% and that of pre-diabetes was 35.7% [8]. Obesity is closely associated with T2D, and among Chinese patients with diabetes, the prevalence of obesity (BMI ≥ 28 kg/m2) is 24.3% [9]. Despite the availability of numerous glucose-lowering drugs, < 20% of Chinese patients with T2D achieve the recommended glycaemic control target (HbA1c < 7%) [7]. Clearly, in China, there is an unmet need for an effective, convenient and easy-to-use treatment option for patients with T2D.
Semaglutide (Ozempic®; Novo Nordisk A/S, Denmark) is a glucagon-like peptide-1 (GLP-1) analogue, approved in numerous countries for the once weekly (OW) subcutaneous (s.c.) treatment of T2D [10, 11]. The large SUSTAIN programme demonstrated that OW s.c. semaglutide provided good glycaemic control versus a wide range of comparators in addition to providing additional clinical benefits such as weight loss, cardiovascular risk reduction and lowering of systolic blood pressure, with a low risk of hypoglycaemia [12,13,14,15,16,17,18,19]. The FDA has recently (January 2020) approved a new indication for Ozempic®: to reduce the risk of major adverse cardiovascular events in adults with T2D and known heart disease [20].
Semaglutide has a long half-life (t½ ≈ 1 week), supporting OW administration [21, 22]. In healthy Caucasian and Japanese subjects, once weekly dosing of semaglutide results in exposure increasing dose proportionally for 0.5 and 1 mg doses [23]. Clinical pharmacology trials and a population pharmacokinetics (PK) analysis of phase 3a trials have shown that dose adjustment is not required by renal or hepatic impairment, race or ethnicity [24,25,26,27]. Furthermore, the population PK analysis showed that the most important predictor of semaglutide exposure is body weight; subjects with higher body weight tend to have lower semaglutide exposure [24, 25]. In addition, drug–drug interaction trials have shown that no dose adjustment is required for commonly used oral medications when administered concomitantly with OW s.c. semaglutide [10, 11, 22, 28].
To further support the approval of OW s.c. semaglutide in China, this clinical pharmacology trial was conducted to evaluate the pharmacokinetics, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects. This trial used the same doses (0.5 and 1.0 mg) and dose-escalation regimen as SUSTAIN CHINA MRCT (ClinicalTrials.gov identifier: NCT03061214), a large multiregional phase 3 clinical trial including approximately 70% subjects from China.
Methods
Trial Design, Subject Eligibility and Dosing
This clinical pharmacology trial was a single-centre, parallel-group, randomised, double-blind, placebo-controlled, multiple-dose PK trial in healthy Chinese subjects.
The trial enrolled subjects at a single site in China (the Clinical Trial Center, Beijing Hospital, Beijing) and was approved by the Ethics Committee of Beijing Hospital. This study was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments, and all subjects provided written informed consent to participate in the study. Eligible subjects were healthy male or female Chinese subjects, aged 18–55 years, with a BMI of 20.0–24.9 kg/m2 and a body weight ≥ 54.0 kg. Subjects with a history of endocrine disorders were excluded, and other exclusion criteria were standard for a clinical pharmacology study in healthy subjects.
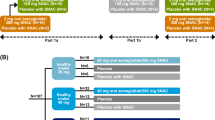
The trial design is shown in Fig. 1. Subjects were randomised 2:1:2:1 to OW s.c. semaglutide 0.5 mg, placebo 0.5 mg, OW s.c. semaglutide 1.0 mg or placebo 1.0 mg. Subjects received trial product at the site by OW s.c. injection in the abdomen on the same weekday for 13 weeks. Doses were escalated every 4 weeks until the target dose was reached, following the FDA- and EMA-approved labels [10, 11].

Trial design. Subjects in blue treatment groups were treated with OW s.c. semaglutide; subjects in grey treatment groups were dosed with corresponding volume of placebo. PK pharmacokinetics, OW once weekly, s.c. subcutaneous
Randomisation and blinding
Randomisation to either active treatment or placebo within each dose level was double-blinded, but dose level (0.5 or 1.0 mg) was open label because of differences in injection volumes. Subjects were assigned to the lowest available randomisation number on the randomisation list and thereby assigned to a treatment group and a subject-specific prepacked trial product (Clinical Supplies Coordination, Novo Nordisk A/S, Søborg). The randomisation list was generated by Novo Nordisk A/S (Søborg), while randomisation and treatment allocation were carried about by the Clinical Pharmacology Research Centre, Beijing Hospital, Beijing. Subjects, trial site staff and sponsor were all blinded to treatment allocation within dose levels.
Trial Endpoints and Safety Assessments
The primary endpoint was area under the semaglutide plasma concentration-time curve over a dosing interval at steady state (AUC0–168 h,SS) after the last administration of OW s.c. semaglutide 0.5 or 1.0 mg.
Secondary PK endpoints for OW s.c. semaglutide 0.5 and 1.0 mg at steady state included maximum concentration (Cmax,SS), time to maximum concentration (tmax,SS), terminal half-life (t½), apparent clearance (CL/FSS), apparent volume of distribution (VSS/F) and trough concentration (Ctrough,SS). Secondary PK endpoints after the first dose of semaglutide (single dose) included AUC0–168 h,SD, Cmax,SD and tmax,SD. The dose-corrected accumulation ratio (Racc,DC) was calculated using AUC0–168 h,SS and AUC0–168 h,SD.
Safety assessments included collection of treatment-emergent adverse events (AEs, defined as any untoward medical occurrence in a subject after the first dose and before the follow-up visit) and technical complaints as well as physical examinations and clinical laboratory tests. The placebo groups were pooled during analysis.
There were no changes to the protocol after trial initiation.
PK Assessments
Blood samples for PK assessment of semaglutide were taken at the following time points after the first dose: 0, 6, 12, 18, 24, 30, 36, 42, 48, 60, 72, 84, 96, 120, 144 and 168 h and at the following time points after the last dose: 0, 6, 12, 18, 24, 30, 36, 42, 48, 60, 72, 84, 96, 120, 144, 168, 336, 504, 672 and 840 h. Additional PK samples were taken before dosing in weeks 4, 8 and 11.
Bioanalysis of blood samples was performed using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) assay [22].
Statistical Analyses
Based on previous data with OW s.c. semaglutide, a sample size of 20 subjects was considered sufficient to achieve an acceptably narrow confidence interval for the dose ratio [22]. Dose proportionality was assessed by the ratio of AUC0–168 h,SS for 0.5 mg/1.0 mg.
To account for potential dropout of up to 20%, a total of 24 subjects were planned for active treatment, and 12 subjects were planned for treatment with placebo (total for both placebo doses), leading to a total number of 36 subjects planned to start in the trial.
The primary endpoint, AUC0–168 h,SS, was derived from the concentration-time curves 0–168 h (1 week) after last semaglutide dose using the non-compartmental, linear trapezoidal method on the observed concentrations using actual time points. The endpoint was analysed by a linear normal model based on the log-transformed values and back-transformed to provide dose ratios alongside 95% confidence intervals. The model included dose group as a fixed factor.
Accumulation ratio (Racc,DC) was calculated as:
where ‘last dose’ is the steady-state dose level of interest (either 0.5 or 1 mg) and ‘first dose’ is the first dose of trial product (0.25 mg).
Analyses were conducted using non-compartmental methods in the statistical software SAS, version 9.4 M5.
Results
Trial Subjects
Subjects were recruited between 21 September 2017 and 23 March 2018. The final follow-up visit for the final patient enrolled was on 7 August 2018. The flow of subjects through the trial is presented in Fig. 2. Two subjects discontinued before trial completion: one subject randomised to OW s.c. semaglutide 1.0 mg was discontinued before the last dose because of ‘use of prescription or non-prescription systemic products or topical medicinal products within 3 weeks prior to the visit' and one subject randomised to placebo was discontinued after the last dose because of a serious AE (traffic accident; considered unrelated to treatment).

Flow diagram of subjects. aOne subject withdrew from the trial, one subject was withdrawn because of violation of a dosing day exclusion criterion (‘use of prescription or non-prescription systemic products or topical medicinal products within 3 weeks prior to the visit’) and four subjects were withdrawn because of adverse events. bVolume-matched placebo (six subjects on placebo 0.5 mg; six subjects on placebo 1.0 mg). cSubject was withdrawn owing to violation of a dosing day exclusion criterion before the final dose. dSubject was withdrawn owing to an SAE unrelated to treatment (traffic accident) after receiving all planned doses of trial product. OW once weekly, SAE serious adverse event, s.c. subcutaneous
The demographics and baseline characteristics were similar across the three treatment arms (Table 1) and reflected the general population of healthy Chinese subjects. All subjects had glycaemic parameters within the normal range at baseline.
Pharmacokinetic Results
The geometric mean plasma concentration of semaglutide is presented over the duration of treatment in Fig. 3a and over a dosing interval (1 week [168 h]) in Fig. 3b. Frequent PK samples were taken after the first dose (day 1) and after the last dose (day 84) of semaglutide, as indicated by the peaks in Fig. 3b. As expected, the mean semaglutide plasma concentration was similar in the semaglutide 0.5 and 1.0 mg groups for the period up until day 56. After day 56, the semaglutide plasma concentration in subjects randomised to semaglutide 1.0 mg increased as these subjects started treatment with a 1.0 mg dose.

Geometric mean semaglutide concentration over time for a the full trial duration and b over a dosing interval at steady state. The dashed lines denote the lower limit of quantification. Values below this limit were imputed. Sema semaglutide
The PK parameters of OW semaglutide at steady state and after the first dose are presented in Table 2. At steady state, the exposure (in terms of AUC0–168 h,SS, Cavg and Cmax,SS) of semaglutide 1.0 mg appeared to be approximately double that of semaglutide 0.5 mg, while clearance and distribution appeared similar for both doses. Both groups were injected with semaglutide 0.25 mg for the first dose, and, as expected, the single dose PK parameters appeared similar for both groups. The dose-corrected accumulation ratio (Racc,DC) was approximately 2, suggesting that, in healthy Chinese subjects, semaglutide accumulated as expected based on the t½ (~1 week) and dosing interval (1 week).
Statistical analysis confirmed that the exposure of s.c. semaglutide increased in a dose-proportional manner in healthy Chinese subjects. The treatment ratio (1.0 mg/0.5 mg) [95% CI] for AUC0–168 h,SS was 1.99 [1.78; 2.23] and the treatment ratio [95% CI] for Cmax,SS was 1.94 [1.74; 2.16].
Exposure in healthy Chinese subjects was slightly higher than that seen in other populations (including healthy European and Japanese subjects and obese European subjects, Fig. 4a); however, after adjusting for differences in body weight, exposure in Chinese subjects was similar to exposure observed in other clinical pharmacology trials (Fig. 4b).

a Average observed semaglutide concentration and b body weight- and dose-normalised average semaglutide concentration across clinical pharmacology trials of OW s.c. semaglutide. Study 1 was conducted in Japan and included healthy Japanese and Caucasian subjects [23]; study 2 was conducted in the UK and included subjects with obesity [29]; studies 3 and 4 were conducted in Germany and included healthy subjects [28]. Cavg was calculated as AUC0–168 h/168 h. Weight- and dose-normalised exposure for each individual subject was calculated as: Cavg/(dose × body weight0.988). Error bars represent 90% ranges for the Cavg
Safety and Tolerability Results
The proportions of subjects reporting AEs were similar for OW s.c. semaglutide 0.5 mg (83%), OW s.c. semaglutide 1.0 mg (92%) and placebo (92%). No new safety signals were identified with OW s.c. semaglutide.
Despite a similar proportion of subjects reporting AEs in all groups, a higher number of AEs were reported with OW s.c. semaglutide 1.0 mg (58 AEs) compared with semaglutide 0.5 mg (44 AEs) and placebo (42 AEs) (Table 3); this was primarily because of a greater number of AEs (e.g. decreased appetite, nausea) in the OW s.c. semaglutide 1.0 mg group. The majority of AEs were mild in severity (Table 3).
There were no deaths in this trial. There was one serious adverse event in the placebo group (traffic accident) that led to discontinuation from the trial after the last dose. This event was considered unrelated to treatment.
Discussion
This trial investigated the PK, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects. Semaglutide exposure increased in a dose-proportional manner for doses of 0.5 and 1.0 mg, and treatment was well tolerated, with no new safety signals identified.
The PK properties of OW semaglutide in healthy Chinese subjects in this trial appeared similar to the PK properties observed in other clinical pharmacology trials with OW semaglutide [23, 28, 29].
The safety and tolerability profile of OW s.c. semaglutide in this trial were consistent with previous trials in healthy subjects [23, 28, 29], and no new safety concerns were identified. Gastrointestinal AEs are a well-established side effect of treatment with GLP-1 receptor agonists [30] and, as expected, gastrointestinal AEs were reported more frequently in subjects treated with OW semaglutide 1.0 mg compared with OW semaglutide 0.5 mg or placebo. However, all gastrointestinal adverse events were mild in severity and none led to treatment discontinuation.
The efficacy and safety of treatment with OW s.c. semaglutide in Chinese subjects with T2D have been demonstrated in a phase 3a clinical trial (SUSTAIN CHINA MRCT, ClinicalTrials.gov identifier: NCT03061214).
Conclusions
This trial demonstrated that the PK, safety and tolerability of OW s.c. semaglutide in healthy Chinese subjects was consistent with previous clinical pharmacology trials of OW s.c. semaglutide in other populations and that treatment with semaglutide was well tolerated in healthy Chinese subjects. The major limitations of this trial are the small sample size and the limited generalisability of the results due to the short duration and the population of healthy subjects.
The results of this trial support that the PK of semaglutide is similar across populations and suggests that no dose adjustment is necessary for semaglutide in Chinese patients with T2D.
References
International Diabetes Federation. IDF Diabetes Atlas, 9th edn. Brussels, Belgium: 2019. 2019 https://www.diabetesatlas.org. Accessed Jan 2020.
Hu C, Jia W. Diabetes in China: epidemiology and genetic risk factors and their clinical utility in personalized medication. Diabetes. 2018;67(1):3–11.
American Diabetes Association. 10. Cardiovascular disease and risk management: standards of medical care in diabetes—2020. Diabetes Care. 2020;43(Supplement 1):S111–34.
Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937–52.
Stark Casagrande S, Fradkin JE, Saydah SH, Rust KF, Cowie CC. The prevalence of meeting A1C, blood pressure, and LDL goals among people with diabetes, 1988–2010. Diabetes Care. 2013;36(8):2271–9.
de Pablos-Velasco P, Parhofer KG, Bradley C, Eschwège E, Gönder-Frederick L, Maheux P, et al. Current level of glycaemic control and its associated factors in patients with type 2 diabetes across Europe: data from the PANORAMA study. Clin Endocrinol (Oxf). 2014;80(1):47–56.
Ji L, Hu D, Pan C, Weng J, Huo Y, Ma C, et al. Primacy of the 3B approach to control risk factors for cardiovascular disease in type 2 diabetes patients. Am J Med. 2013;126(10):925e11–22.
Wang L, Gao P, Zhang M, Huang Z, Zhang D, Deng Q, et al. Prevalence and ethnic pattern of diabetes and prediabetes in China in 2013. JAMA. 2017;317(24):2515–23.
Hou X, Lu J, Weng J, Ji L, Shan Z, Liu J, et al. Impact of waist circumference and body mass index on risk of cardiometabolic disorder and cardiovascular disease in Chinese adults: a national diabetes and metabolic disorders survey. PLoS ONE. 2013;8(3):e57319.
Novo Nordisk. Ozempic (semaglutide) injection, for subcutaneous use—highlights of prescribing information 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209637lbl.pdf. Accessed Jan 2020.
Novo Nordisk A/S. Ozempic summary of product characteristics 2018. https://www.ema.europa.eu/en/documents/product-information/ozempic-epar-product-information_en.pdf. Accessed Jan 2020.
Sorli C, Harashima SI, Tsoukas GM, Unger J, Karsbol JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(4):251–60.
Ahrén B, Masmiquel L, Kumar H, Sargin M, Derving Karsbøl J, Hald Jacobsen S, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017;5(5):351–4.
Ahmann AJ, Capehorn M, Charpentier G, Dotta F, Henkel E, Lingvay I, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018;41(2):258–66.
Aroda VR, Bain SC, Cariou B, Piletic M, Rose L, Axelsen M, et al. Efficacy and safety of once-weekly semaglutide versus once-daily insulin glargine as add-on to metformin (with or without sulfonylureas) in insulin-naive patients with type 2 diabetes (SUSTAIN 4): a randomised, open-label, parallel-group, multicentre, multinational, phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(5):355–66.
Rodbard HW, Lingvay I, Reed J, de la Rosa R, Rose L, Sugimoto D, et al. Semaglutide added to basal insulin in type 2 diabetes (SUSTAIN 5): a randomized, controlled trial. J Clin Endocrinol Metab. 2018;103(6):2291–301.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375(19):1834–44.
Kaneko S, Nishijima K, Bosch-Traberg H, Kaku K, Seino Y. Efficacy and safety of adding liraglutide to existing insulin regimens in Japanese patients with type 2 diabetes mellitus: a post-hoc analysis of a phase 3 randomized clinical trial. J Diabetes Investig. 2018;9(4):840–9.
Kaku K, Yamada Y, Watada H, Abiko A, Nishida T, Zacho J, et al. Safety and efficacy of once-weekly semaglutide vs additional oral antidiabetic drugs in Japanese people with inadequately controlled type 2 diabetes: a randomized trial. Diabetes Obes Metab. 2018;20(5):1202–12.
Novo Nordisk A/S. FDA approves Ozempic® for cardiovascular risk reduction in adults with type 2 diabetes and known heart disease, updates Rybelsus® label [Internet] NovoNordisk-us.com 2020. https://www.novonordisk-us.com/media/news-releases.html?122981. Accessed Jan 2020.
Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–80.
Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55(5):497–504.
Ikushima I, Jensen L, Flint A, Nishida T, Zacho J, Irie S. A randomized trial investigating the pharmacokinetics, pharmacodynamics, and safety of subcutaneous semaglutide once-weekly in healthy male Japanese and Caucasian subjects. Adv Ther. 2018;35(4):531–44.
Overgaard RV, Delff PH, Petri KCC, Anderson TW, Flint A, Ingwersen SH. Population pharmacokinetics of semaglutide for type 2 diabetes. Diabetes Ther. 2019;10(2):649–62.
Carlsson Petri KC, Ingwersen SH, Flint A, Zacho J, Overgaard RV. Semaglutide s.c. once-weekly in type 2 diabetes: a population pharmacokinetic analysis. Diabetes Ther. 2018;9(4):1533–47.
Marbury TC, Flint A, Jacobsen JB, Derving Karsbol J, Lasseter K. Pharmacokinetics and tolerability of a single dose of semaglutide, a human glucagon-like peptide-1 analog, in subjects with and without renal impairment. Clin Pharmacokinet. 2017;56(11):1381–90.
Jensen L, Kupcova V, Arold G, Pettersson J, Hjerpsted J. Pharmacokinetics and tolerability of semaglutide in people with hepatic impairment. Diabetes Obes Metab. 2018;20(4):977–86.
Hausner H, Derving Karsbol J, Holst AG, Jacobsen JB, Wagner FD, Golor G, et al. Effect of semaglutide on the pharmacokinetics of metformin, warfarin, atorvastatin and digoxin in healthy subjects. Clin Pharmacokinet. 2017;56(11):1391–401.
Blundell J, Finlayson G, Axelsen M, Flint A, Gibbons C, Kvist T, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. 2017;19(9):1242–51.
Horowitz M, Aroda VR, Han J, Hardy E, Rayner CK. Upper and/or lower gastrointestinal adverse events with glucagon-like peptide-1 receptor agonists: incidence and consequences. Diabetes Obes Metab. 2017;19(5):672–81.
Acknowledgements
We thank Umut Erhan, Azadeh Houshman-Øregaard and Anders Strathe of Novo Nordisk A/S (Denmark) and Zu Ning of Novo Nordisk China A/S (China) for their contributions to the manuscript.
Funding
This study, the Rapid Service and Open Access Fees (ClinicalTrials.gov: NCT03288740) were funded by Novo Nordisk A/S, Denmark.
Medical Writing and Editorial Assistance
We thank Christopher W. Williams of Novo Nordisk A/S (Denmark) for medical writing support and Fraser Harris, MRes, of AXON Communications for editorial and submission support, funded by Novo Nordisk.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Prior Presentation
This study has been presented as a poster at the 80th Scientific Sessions of the American Diabetes Association, June 12–16, 2020.
Disclosures
Aixin Shi, Panpan Xie and Xuemei He have nothing to disclose. Lasse Lykke Nielsen, Sine Pfeiffer Haugaard and Trine Vang Skjøth are Novo Nordisk employees and shareholders.
Compliance with Ethics Guidelines
The trial enrolled subjects at a single site in China (the Clinical Trial Center, Beijing Hospital, Beijing) and was approved by the Ethics Committee of Beijing Hospital. This study was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments, and all subjects provided written informed consent to participate in the study.
Data Availability
The datasets generated during and/or analysed during the current study are available on reasonable request. The access request proposal form and the access criteria can be found at www.novonordisk-trials.com.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shi, A., Xie, P., Nielsen, L.L. et al. Pharmacokinetics, Safety and Tolerability of Once-Weekly Subcutaneous Semaglutide in Healthy Chinese Subjects: A Double-Blind, Phase 1, Randomized Controlled Trial. Adv Ther 38, 550–561 (2021). https://doi.org/10.1007/s12325-020-01548-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01548-y