
Abstract
Recent genetic and preclinical studies have increased our understanding of the immunopathogenesis of alopecia areata (AA). This has allowed expedited development of targeted therapies for the treatment of AA, and a paradigm shift in our approach and understanding of autoimmunity and the hair follicle. The synergy between preclinical studies, animal models, and translational studies has led to unprecedented advances in the treatment options for AA, ultimately benefiting patients who have had little recourse. In this review, we summarize the scientific field of contemporary AA research, and look forward to potential new technologies and developments.
Similar content being viewed by others

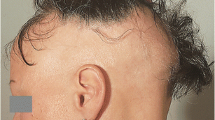

Introduction
Alopecia areata (AA) is one of the most prevalent autoimmune diseases in the USA, affecting approximately 5.3 million people, including both sexes across all ethnicities, with a lifetime risk of 1.7% [1]. This is comparable to the estimated 2% lifetime incidence of AA globally, which comes with a significant healthcare burden [2]. Despite this high prevalence, there have been no evidence-based treatments for AA as recently as 5 years ago. Recent scientific and clinical efforts have uncovered novel insights into the pathogenesis of AA, which have led to the development of new translational treatments. This recent progress has revived the “translational landscape” of AA, in terms of its etiology, treatment options, and prognosis, and has given new hope to the millions of patients suffering from the disease. In this brief review, we will survey the rapidly evolving landscape of AA, and provide an overview of how basic research, in collaboration with clinical dermatology, has transformed the future prospects for treatment of this devastating and highly prevalent disease. Treatment algorithms are out of the scope of this review, and will be discussed elsewhere. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Pathogenesis
Historically, AA has been classified as an autoimmune disease of the hair follicle due to the characteristic T cell accumulation at the hair bulb, also known as the histological “swarm of bees”. However, a detailed description of this immune infiltrate remained unexplored. Little was known about the initial trigger, factors required for maintenance, and identity of the elusive autoantigen(s). Recent fundamental and basic science studies have greatly improved our understanding of AA and its possible causes.
Genetic Basis of AA
In the past decade, genome-wide association studies (GWAS) and genetic linkage studies have been central to the elucidation of the pathogenesis of AA. Compared to GWAS of other autoimmune and immune-mediated diseases, the “hits” obtained from the AA GWAS have yielded a higher number of meaningful candidate genes that have shed light on the mechanisms of disease in AA [3], providing new therapeutic targets.
In our first GWAS for AA, which included 1054 AA patients from a comprehensive registry (National Alopecia Areata Registry) in North America, we identified eight regions of the genome that were significantly associated with AA [4]. Like other autoimmune conditions, the human leukocyte antigen (HLA) locus was significantly represented in the AA GWAS, supporting previous candidate gene association studies [5, 6]. Interestingly, the second highest peak uncovered in our GWAS, and confirmed in other cohorts, mapped to the ULBP6/ULBP3 gene locus on chromosome 6q. Functional studies confirmed that ULBP3 was aberrantly upregulated as a danger signal on the hair follicles of AA patients [4, 7], which contributed to the collapse of immune privilege, and the recruitment of cytotoxic CD8+ NKG2D+ T cells to carry out destruction of the hair follicle. We subsequently showed that this subset of T cells was both necessary and sufficient for the development of AA. Meta-analysis of GWAS studies in AA was conducted on the combined North American and European AA cohorts [8], and confirmed the significance of these susceptibility loci, as well as identifying several new loci.
Applying pathway and network analyses (e.g., Gene Ontology term enrichment and proteomic interactions) to the GWAS data has also uncovered sets of genes that contribute to pathogenic processes. This type of analysis grouped AA with other autoimmune diseases like type 1 diabetes, rheumatoid arthritis (RA), and celiac disease. Pathways that are implicated in the pathogenesis of this group of autoimmune diseases include antigen processing and presentation, co-stimulatory pathways, and JAK-STAT signaling [9]. These associations led to a re-analysis of the underlying immunopathogenesis of AA, and to a consideration of employing new therapies that target these pathways specifically.
Immunopathogenesis
Adaptive immune responses were traditionally classified into Th1 and Th2 responses, depending on the components of the cytokine milieu in the diseased state. This binary view of the immune response has been shown to be insufficient to account for the complex immune profiles of many diseases. Historically, AA was assigned to the Th1 category owing to its significant interferon-gamma (IFN-γ) signature. However, AA has also been epidemiologically associated with atopy [10], a classic Th2 condition defined by a predominance of IL-4 and IL-13. While measuring cytokine transcripts in circulating immune cells in AA tend to favor a Th1 profile [11, 12], lesional skin in AA exhibited a more mixed profile [13]. Gene expression profiles of AA lesional skin have also uncovered mixed immune response signatures [14]. This intriguing finding suggests that the immune response in AA may be heterogenous, and may fluctuate and evolve within a single patient through time, or may even signal distinct disease subtypes that may require different therapeutic approaches.
Autoantigens of the Hair Follicle
A few autoimmune diseases (i.e., autoimmune thyroiditis, celiac disease, rheumatoid arthritis) have well-characterized autoantigens that are the known targets for the immune system. Most other autoimmune conditions and immune-mediated diseases, including AA and psoriasis, do not yet have any autoantigen that is clearly associated with their pathogenesis. The hair follicle is regarded as a relatively immune-privileged site [15], with generally low expression of MHC class I molecules and “danger signals” that are overexpressed in disease (e.g., ULBP6/3). In order to sequester hair follicle antigens from the immune system, particularly during the hair cycle when significant tissue remodeling occurs, the hair follicle microenvironment needs to be precisely regulated. Some studies have suggested that failure of this mechanism leads to a “collapse of immune privilege” and sensitization of circulating T cells to keratinocyte and/or melanocyte peptides, which might precipitate AA [16]. Autoantibodies specific for hair follicle proteins such as trichohyalin and certain keratins have been detected in both mouse and human cases of AA [17, 18], but more work remains to expand the repertoire of discovered AA autoantigens and their relevance to disease pathogenesis.
Preclinical Studies and Animal Models
Basic science and preclinical research are not frequently appreciated when discussing the clinical aspects of a disease, but nonetheless they provide the bedrock of innovation that drives the technology and progress that fuels novel therapies.
Animal Model of AA
A well-validated animal model can make a significant impact in the study of human disease. In the case of AA, the C3H/HeJ mouse was found to spontaneously develop an immune-mediated hair loss that resembled human AA both histopathologically and immunologically, albeit at a low frequency, as the mice age [19]. This phenotype was found to be transferrable with skin grafts from an affected mouse to younger, congenic recipients [20]. This model revolutionized the field of AA research by establishing a reliable animal model that closely recapitulated the human disease. Recently, this process has been reproduced with transfer of skin-draining lymph node cells, abrogating the need for invasive surgical procedures [21].
Humanized models of AA have also been attempted. By grafting normal human scalp skin onto immunocompromised mice, and transferring peripheral blood monocytes from an unrelated healthy donor enriched for the pathogenic NKG2D+ population, one can precipitate an autoimmune type hair loss which may have similarities to AA, although the tissues are derived from normal healthy donors [22].
Targeting Effector Cytokines/Chemokines in Animal Models of AA
As knowledge accumulates on the immunopathogenesis of AA, researchers have been able to systematically target and investigate the roles of upstream and downstream effectors of the system. The identification of the pathogenic subset of NKG2D+ CD8+ cytotoxic T cells led to the characterization of the cytokine milieu that they secrete. This subset of T cells was found to respond to IFN-induced chemokines CXCL9/10/11, released by the hair follicle itself. Antagonism of their common chemokine receptor (CXCR3) on T cells was subsequently shown to be sufficient to prevent onset of AA in a mouse model [23].
Diagnosis and Disease Prognostication
AA is normally a straightforward clinical diagnosis to make, particularly when it presents in its classical form with focal or multifocal patches of acute non-scarring alopecia, and a positive hair-pull test. Occasionally, a skin biopsy might be required to distinguish diffuse AA from other conditions such as female androgenetic alopecia or telogen effluvium. In these cases, AA is unequivocally associated with the histopathological finding of a peribulbar immune infiltrate of cytotoxic T cells (the so-called swarm of bees). However, newer non-invasive technologies discussed below have been adopted to facilitate in diagnosis of difficult cases.
Trichoscopy and Reflectance Confocal Microscopy
Dermatoscopy has been an indispensable tool for the management of melanocytic lesions, and training to detect melanoma amongst dermatology residents has reached a very high standard. Thus, most, if not all, dermatologists are equipped with a state-of-the-art dermatoscope, which has recently been adopted for diagnosing hair disorders [24]. In addition to the classical “exclamation mark hairs” which are found at the periphery of AA lesions, dermatoscopic, or trichoscopic features of AA include yellow dots, which correlate with dystrophic follicular epithelium and sebaceous glands [25]. Digital videodermatoscopy has also been employed for the diagnosis and follow-up of hair disorders in many clinics. Reflectance confocal microscopy is another tool that may be employed for non-invasive diagnosis of AA, with the “yellow dots” clearly visible as degenerated follicles and, in some cases, even the immune infiltrate can be visualized [26].
Stratification of Patients with ALADIN Gene Expression Profiling
The era of personalized medicine has brought with it a vast body of large datasets related to sequencing, genotyping, and gene expression. Not only do we have information from GWAS hits that may suggest potential new druggable targets but RNA-Seq and microarray data from individual patients are beginning to provide insight into the molecular differences in disease pathogenesis from patient to patient. Integrating the relative expression of genes in the interferon (IFN), cytotoxic T cell (CTL), and keratinocyte (KER) clusters, a new metric known as the Alopecia Areata Disease Activity Index (ALADIN) has been developed [14]. ALADIN is specific to AA, and correlates with disease activity, making it a suitable biomarker for assessment of clinical response to treatment, and to identify patients who may or may not respond to certain therapeutic modalities [14].
Treatment
Treatment modalities for AA are diverse and to date have not been reliably effective. Until recently, the treatments for AA have been generally non-specific and employ a “scorched earth” strategy of either suppressing (intralesional and topical corticosteroids) or stimulating (topical immunotherapy, excimer laser therapy) the immune system, with varying results. Very few well-designed randomized controlled clinical trials have been conducted to justify their widespread adoption. These have been reviewed extensively elsewhere [27, 28]. For this review, we will focus on the innovative new therapies that have been developed for the treatment of AA.
Janus Kinase Inhibitors
A significant breakthrough in the treatment of AA has been the discovery of the importance of the JAK-STAT pathway in the initiation and the maintenance of the diseased state (see “Pathogenesis”). This provided the rationale to use small molecule JAK inhibitors, which have been FDA-approved for other diseases (tofacitinib for rheumatoid arthritis and ruxolitinib for myelofibrosis), for the treatment of AA. Treatment-resistant patients with extensive disease, alopecia universalis or totalis, were found in open-label trials to respond with full regrowth of hair after a twice-daily regimen of ruxolitinib within 3–5 months. This effect was supported by case reports that reported JAK inhibitors reversing co-incidental AA in patients being treated for other diseases [29, 30]. Further clinical trials are underway for systemic tofacitinib and ruxolitinib for the treatment of AA [31, 32], as well as topical formulations of the JAK inhibitors.
Co-stimulatory Blockade
Cytotoxic T lymphocyte antigen (CTLA) 4 is a negative regulator of T cells, attenuating the immune response by blocking co-stimulatory signals by interfering with the interaction between CD28 on T cells and CD80/CD86 on antigen-presenting cells. Its dysfunction has been associated with several autoimmune and immune-mediated conditions like thyroiditis [33], rheumatoid arthritis [34], and inflammatory bowel disease [35]. Single nucleotide polymorphisms in the CTLA4 gene have also been associated with AA in some populations [36]. Its location on chromosome 2q33.2 was also among the GWAS hits for AA [4]. Abatacept, a recombinant fusion protein (CTLA4-Ig), has been an effective and successful treatment for RA and other autoimmune diseases [37]. Preclinical studies have also shown that abatacept is effective in preventing the mouse model of AA if given before the onset of disease [38], providing rationale and promise for its efficacy in human AA patients. Clinical trials are underway to investigate its role in AA (ClinicalTrials.gov NCT02018042).
Simvastatin/Ezetimibe
Several case reports [39, 40] and a small case–control study [41] have suggested that simvastatin/ezetimibe, a common treatment for hyperlipidemia, may have immunomodulatory effects that might benefit AA patients. Considering atherosclerosis to be a disease of the adaptive immune system [42], this drug combination has been postulated to have effects on antigen presentation, lymphocyte trafficking, and regulatory T cell induction. Inevitably, clinicians will be testing this treatment off-label on their most recalcitrant cases of AA, since simvastatin/ezetimibe has an acceptable safety record. Larger randomized controlled studies are required to confirm its efficacy.
Biologic Therapies for AA
Similar preclinical studies are directed at other pathways that intersect with the pathogenic NKG2D+ CD8+ T cell subset, one of which may eventually translate into the clinical realm. Since anti-TNF (tumor necrosis factor) therapies have uniformly failed in AA [43] and have been reported to precipitate AA in some patients [44], there has been a resurgence of interest in testing other biological modalities in the setting of AA. Preclinical studies have established the roles of IL-15 [45], IFN-γ [46], and IL-2 [47] in the pathogenesis of AA in mice, and specific therapies targeting these pathways are being investigated. Low-dose IL-2, which has been used to stimulate expansion of regulatory T cells in the treatment of autoimmune disease such as type 1 diabetes [48], has been used in AA with modest results [49].
As alluded to before, the apparent heterogeneity of the immune response in AA has led to speculation that Th2 and even Th17 immune pathways may be involved in the development of the disease. This stems from gene expression data and cytokine profiling of a small and probably heterogenous group of patients [13]. A subsequent small case series showed that patients with a higher inflammatory score measured with ALADIN responded well to ustekinumab [50], a humanized monoclonal antibody to the p40 subunit that is central to the IL-12/IL-23 signaling pathways of the Th17 response. Further work to deconstruct the components of the immune response in AA to determine the heterogeneity and spectrum of disease phenotypes that may present in different patients will help to tailor treatment strategies in a precise manner. This personalized approach will maximize the efficacy of treatment, while minimizing the potential adverse effects and drug exposures.
Conclusions
As our knowledge and understanding of the immune system progress, we have been able to dissect and target increasingly precise components of the immune response. With this understanding also comes the revelation that the complexities of the immune system cannot be explained by simple dichotomies, and that every patient is different, and requires a precise, personalized treatment. Our conceptual understanding of autoimmunity, illustrated here with the case of AA, is evolving to encompass new concepts such that the entire approach of diagnosis, prognosis, and management is evolving along with it. The coming years will herald new exciting therapies for our patients, targeting pathways that we may not even have suspected previously, which will in turn bring us more questions and new perceptions of this common, devastating disease.
References
Safavi KH, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70(7):628–33.
Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397–403.
Petukhova L, Christiano AM. The genetic architecture of alopecia areata. J Investig Dermatol Symp Proc. 2013;16(1):S16–22.
Petukhova L, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466(7302):113–7.
Colombe BW, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5 Pt 1):757–64.
Barahmani N, et al. Major histocompatibility complex class I chain-related gene A polymorphisms and extended haplotypes are associated with familial alopecia areata. J Invest Dermatol. 2006;126(1):74–8.
Moftah NH, et al. ULBP3: a marker for alopecia areata incognita. Arch Dermatol Res. 2016;308(6):415–21.
Betz RC, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun. 2015;6:5966.
Petukhova L, Christiano AM. Functional interpretation of genome-wide association study evidence in alopecia areata. J Invest Dermatol. 2016;136(1):314–7.
Penders AJ. Alopecia areata and atopy. Dermatologica. 1968;136(5):395–9.
Kuwano Y, et al. Serum chemokine profiles in patients with alopecia areata. Br J Dermatol. 2007;157(3):466–73.
Sadeghi S, et al. Study of Th1/Th2 balance in peripheral blood mononuclear cells of patients with alopecia areata. Acta Microbiol Immunol Hung. 2015;62(3):275–85.
Suarez-Farinas M, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J Allergy Clin Immunol. 2015;136(5):1277–87.
Jabbari A, et al. Molecular signatures define alopecia areata subtypes and transcriptional biomarkers. EBioMedicine. 2016;7:240–7.
Paus R, Bertolini M. The role of hair follicle immune privilege collapse in alopecia areata: status and perspectives. J Investig Dermatol Symp Proc. 2013;16(1):S25–7.
Wang EH, et al. Identification of autoantigen epitopes in alopecia areata. J Invest Dermatol. 2016;136(8):1617–26.
Tobin DJ, et al. Autoantibodies to hair follicles in C3H/HeJ mice with alopecia areata-like hair loss. J Invest Dermatol. 1997;109(3):329–33.
Leung MC, et al. Trichohyalin is a potential major autoantigen in human alopecia areata. J Proteome Res. 2010;9(10):5153–63.
Sundberg JP, et al. C3H/HeJ mouse model for alopecia areata. J Invest Dermatol. 1995;104(5 Suppl):16S–7S.
McElwee KJ, et al. Experimental induction of alopecia areata-like hair loss in C3H/HeJ mice using full-thickness skin grafts. J Invest Dermatol. 1998;111(5):797–803.
Wang EH, et al. Transfer of alopecia areata to C3H/HeJ mice using cultured lymph node-derived cells. J Invest Dermatol. 2015;135(10):2530–2.
Gilhar A, Keren A, Paus R. A new humanized mouse model for alopecia areata. J Investig Dermatol Symp Proc. 2013;16(1):S37–8.
Dai Z, et al. CXCR3 blockade inhibits T cell migration into the skin and prevents development of alopecia areata. J Immunol. 2016;197(4):1089–99.
Estefan J, et al. Alopecia areata–Part II: diagnosis and pathology. Skinmed. 2015;13(2):121–6.
Tosti A, et al. The role of scalp dermoscopy in the diagnosis of alopecia areata incognita. J Am Acad Dermatol. 2008;59(1):64–7.
Ardigo M, et al. Reflectance confocal microscopy of the yellow dot pattern in alopecia areata. Arch Dermatol. 2011;147(1):61–4.
Hordinsky M, Donati A. Alopecia areata: an evidence-based treatment update. Am J Clin Dermatol. 2014;15(3):231–46.
Hordinsky MK. Current treatments for alopecia areata. J Investig Dermatol Symp Proc. 2015;17(2):44–6.
Pieri L, Guglielmelli P, Vannucchi AM. Ruxolitinib-induced reversal of alopecia universalis in a patient with essential thrombocythemia. Am J Hematol. 2015;90(1):82–3.
Higgins E, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. 2015;135(2):551–3.
Kennedy Crispin M, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1(15):e89776.
Mackay-Wiggan J, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. 2016;1(15):e89790.
Patel H, et al. Association of cytotoxic T-lymphocyte antigen 4 (CTLA4) and thyroglobulin (TG) genetic variants with autoimmune hypothyroidism. PLoS One. 2016;11(3):e0149441.
Benhatchi K, et al. CTLA4 exon1 A49G polymorphism in Slovak patients with rheumatoid arthritis and Hashimoto thyroiditis-results and the review of the literature. Clin Rheumatol. 2011;30(10):1319–24.
Repnik K, Potocnik U. CTLA4 CT60 single-nucleotide polymorphism is associated with Slovenian inflammatory bowel disease patients and regulates expression of CTLA4 isoforms. DNA Cell Biol. 2010;29(10):603–10.
Megiorni F, et al. Cytotoxic T-lymphocyte antigen 4 (CTLA4) +49AG and CT60 gene polymorphisms in alopecia areata: a case-control association study in the Italian population. Arch Dermatol Res. 2013;305(7):665–70.
Nusslein HG, et al. Efficacy and prognostic factors of treatment retention with intravenous abatacept for rheumatoid arthritis: 24-month results from an international, prospective, real-world study. Clin Exp Rheumatol. 2016;34(3):489–99.
Petukhova L, et al. The genetics of alopecia areata: what’s new and how will it help our patients? Dermatol Ther. 2011;24(3):326–36.
Ali A, Martin JMT. Hair growth in patients alopecia areata totalis after treatment with simvastatin and ezetimibe. J Drugs Dermatol. 2010;9(1):62–4.
Lattouf C, et al. Treatment of alopecia areata with simvastatin/ezetimibe. J Am Acad Dermatol. 2015;72(2):359–61.
Loi C, Starace M, Piraccini BM. Alopecia areata (AA) and treatment with simvastatin/ezetimibe: experience of 20 patients. J Am Acad Dermatol. 2016;74(5):e99–100.
Moreira FT, et al. Effects of two lipid lowering therapies on immune responses in hyperlipidemic subjects. Life Sci. 2014;98(2):83–7.
Bolduc C, Bissonnette R. Safety and efficacy of adalimumab for the treatment of severe alopecia areata: case series of three patients. J Cutan Med Surg. 2012;16(4):257–60.
Hernandez MV, et al. Development of alopecia areata after biological therapy with TNF-alpha blockers: description of a case and review of the literature. Clin Exp Rheumatol. 2009;27(5):892–3.
Xing L, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20(9):1043–9.
Freyschmidt-Paul P, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155(3):515–21.
Freyschmidt-Paul P, et al. The functional relevance of the type 1 cytokines IFN-gamma and IL-2 in alopecia areata of C3H/HeJ mice. J Investig Dermatol Symp Proc. 2005;10(3):282–3.
Pham MN, von Herrath MG, Vela JL. Antigen-specific regulatory T cells and low dose of IL-2 in treatment of type 1 diabetes. Front Immunol. 2015;6:651.
Castela E, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol. 2014;150(7):748–51.
Guttman-Yassky E, et al. Extensive alopecia areata is reversed by IL-12/IL-23p40 cytokine antagonism. J Allergy Clin Immunol. 2016;137(1):301–4.
Acknowledgements
This work was funded by NIH Grant P50 AR070588-01. No sponsorship was received for the article processing charges. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Dr Christiano is a consultant to Aclaris Therapeutics. Dr Wang has nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/C818F0606DABA121.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, E.C.E., Christiano, A.M. The Changing Landscape of Alopecia Areata: The Translational Landscape. Adv Ther 34, 1586–1593 (2017). https://doi.org/10.1007/s12325-017-0540-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0540-9