
Abstract
The bone marrow constitutes an unique microenvironment for cancer cells in three specific aspects. First, the bone marrow actively recruits circulating tumor cells where they find a sanctuary rich in growth factors and cytokines that promote their proliferation and survival. When in the bone marrow, tumor cells profoundly affect the homeostasis of the bone and the balance between osteogenesis and osteolysis. As a consequence, growth and survival factors normally sequestered into the bone matrix are released, further fueling cancer progression. Second, tumor cells actively recruit bone marrow-derived precursor cells into their own microenvironment. When in the tumors, these bone marrow-derived cells contribute to an inflammatory reaction and to the formation of the tumor vasculature. Third, bone marrow-derived cells can home in distant organs, where they form niches that attract circulating tumor cells. Our understanding of the contribution of the bone marrow microenvironment to cancer progression has therefore dramatically improved over the last few years. The importance of this new knowledge cannot be underestimated considering that the vast majority of cancer treatments such as cytotoxic and myeloablative chemotherapy, bone marrow transplantation and radiation therapy inflict a trauma to the bone marrow microenvironment. How such trauma affects the influence that the bone marrow microenvironment exerts on cancer is still poorly understood. In this article, the reciprocal relationship between the bone marrow microenvironment and tumor cells is reviewed, and its potential impact on cancer therapy is discussed.
Similar content being viewed by others


Introduction
In their seminal paper published in 2000, Hanahan and Weinberg [1] identified six critical hallmarks of cancer that together represent the fundamental mechanisms underlying malignant transformation and cancer progression. In view of the complexity of the mechanisms underlying each of these hallmarks, it is not surprising that cancer remains the leading cause of death in humans age 40 to 65 and a major challenge for the scientific and medical community. There is a fundamental difference among these hallmarks. Self sufficiency in growth signals, the ability to evade apoptosis, insensitivity to anti-growth signals and limitless replicative potential are four among the six hallmarks that have their basis on genetic and epigenetic changes in the genome of the cancer cells. In contrast, sustained angiogenesis and the capability to invade and metastasize, are two hallmarks that have their basis not only on genetic and epigenetic alterations but also on dramatic changes in normal cellular and acellular elements that support malignant cells. These non-malignant components of the tumor are known to form the tumor microenvironment. For the last two decades, the primary focus of investigations on the microenvironment has been on the tissues that are directly adjacent to primary and metastatic tumors. Connective tissue proteins, the basement membrane, adjacent endothelial cells (EC), and stromal fibroblasts have been the subject of numerous investigations in tumor invasion and angiogenesis that have led to the recent successful use of anti-angiogenic therapies [2]. In this aspect, the mechanisms by which the bone marrow and the bone, which are frequent sites of metastasis, provide a unique microenvironment for the proliferation and survival of metastatic tumor cells have been well characterized [3, 4]. However, the fact that non-malignant cells from distant organs, primarily the bone marrow, can contribute to the microenvironment of primary and metastatic tumors has received little attention in the past but has now been increasingly appreciated. Tumors actively recruit bone marrow-derived cells (BMDC) which contribute to inflammation and vascularization in the tumor microenvironment (Table 1). Central to the contribution of the bone marrow to tumor progression are bone marrow-derived stem cells (BMDSC). Evidence supporting a contribution of these cells to tumor progression and distant metastasis has now been clearly obtained from animal experiments in which lethally irradiated recipient mice are transplanted with donor bone marrow cells that express a marker protein such as green fluorescent protein (GFP), luciferase or LacZ.
The relationship between the bone marrow and tumor cells is reciprocal. Tumor cells leave the primary tumor as they intravasate into the blood circulation and are actively recruited into the bone marrow microenvironment. When homed to the bone marrow they disrupt the homeostatic balance between osteogenesis and osteolysis and create a microenvironment that favors their growth and survival [5]. And vice-versa, BMDC leave the bone marrow osteoblastic niche to enter the blood circulation and colonize the primary tumor where they contribute to the tumor vasculature and to an inflammatory reaction that deeply affects malignant transformation and progression [6, 7]. BMDC can also colonize distant organs and form a pre-metastatic niche that will attract circulating tumor cells and promote the formation of macroscopic tumors [8]. In this article, these aspects will be specifically discussed. The bone marrow is also home to non-innate immunocompetent cells (cytotoxic T cells, NK cells, antigen presenting cells and B cells) that initiate and coordinate an immune attack against cancer cells. The contribution of these cells to cancer progression will however not be discussed in this article as it is beyond its scope
Bone Marrow-Derived Stem Cells
Hematopoietic Stem Cells and Endothelial Progenitor Cells
Bone marrow-derived hematopoietic stem cells (HSC) are developmentally related to endothelial progenitors and arise during embryonic development from a common hemangioblast precursor cell [9]. Embryonic hemangioblasts are phenotypically similar to adult HSC and express c-Kit (CD117), Sca-1 and CD133, but not Lin [10]. These cells have been shown to be a common precursor of both hematopoietic and endothelial cells [10, 11]. As they cells mature they express one of two forms of the vascular endothelial cell growth factor receptor (VEGFR)-1 or -2 [7, 12]. HSC express VEGFR-1 as they proliferate, differentiate, and migrate in the blood circulation. These cells, which are CD45 positive, colonize the primary tumor where they contribute to inflammation and to the perivascular space. They also colonize distant organs where they contribute to metastasis as will be discussed later. In contrast, cells expressing VEGFR-2 differentiate into circulating endothelial progenitor cells (CEP) and later mature EC that merge with EC originating from adjacent blood vessels to form the tumor vasculature [13, 14]. CEP in the peripheral blood, which derive from hemangioblasts, are phenotypically defined as negative for CD45 and positive for CD13/aminopeptidase N, VEGFR-2, CD117/Kit and CD34 [15]. Since CD34 is also expressed on mature circulating EC (CEC), the expression of the marker CD133 allows the distinction between CEP which are negative for CD133 and CEC that express CD133 [16]. However, some studies have shown that HSC are not the only source of bone marrow derived EC [17], suggesting therefore that endothelial lineage probably derives from hemangioblasts and non-hematopoietic stem cells in adults.
Mesenchymal Stem Cells
The second type of stem cells in the bone marrow is the pool of mesenchymal stem cells (MSC) [18]. These cells do not express the HSC marker CD34, but express multiple other markers like Stro-1, CD105/endoglin, CD44/hyaluronic receptor and CD90 [19, 20]. When cultured, they typically adhere to plastic or ECM proteins, and can differentiate into a large variety of mature mesenchymal cells including osteoblasts/osteocytes, chondrocytes, cardiomyocytes, myocytes, adipocytes and even neuronal cells [21, 22]. Their pool in the bone marrow is enhanced by Wnt-3a, a member of the Wnt family of growth factors. [23]. These cells are the subject of a growing attention because of their potential therapeutic value in wound repair and tissue regeneration. Their contribution to tumor progression is less well understood than HSC but they play an important role in bone metastasis, as will be discussed later.
Bone Marrow Niches
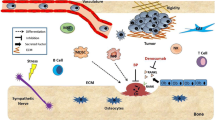
The fate of BMDSC, in particular HSC, is controlled by the microenvironment in the bone marrow. This microenvironment is made of specific niches that provide support for the proliferation and maintenance of stem cells [24]. Interactions between stem cells and their microenvironment regulate their maintenance, proliferation, differentiation and migration into the blood circulation (Fig. 1). Distinct niches have been anatomically and physiologically defined within the bone marrow [25, 26]. In the endosteal region, osteoblasts and other mesenchymal-derived stromal cells such as reticular cells, fibroblasts and adipocytes constitute the osteoblastic niche that supports the maintenance of HSC in a quiescent and undifferentiated state. Alteration of normal osteoblastic function results in a decrease of hematopoiesis and vice versa expansion of osteoblasts is associated with an increased number of HSC in the bone marrow [27, 28]. In the central region of the bone marrow, EC in sinusoids contribute to the formation of a second compartment defined as the vascular niche. BMDSC are recruited in this niche where they proliferate, differentiate and migrate through the sinusoidal wall [26]. Thus whereas the osteoblastic niche promotes the maintenance of a pool of undifferentiated stem cells, the vascular niche promotes the release of differentiated and mature hematopoietic cells.

VEGFR-1 and 2 positive precursor cells are released from the bone marrow. HSC are retained in the endosteal niche by a combination of cell–cell (Tie-2/angio-1), cell-matrix (α4β1/osteopontin) and receptor–ligand (CXCR-4/SDF-1) interactions. The mobilization of these cells toward the vascular niche and their release in the peripheral blood is in part controlled by MMP-9 which (1) degrades SDF-1, (2) degrades osteopontin, (3) solubilizes cKit-L and (4) degrades the basement membrane
The mechanisms by which the microenvironment of the niche interacts with stem cells and controls their fate include adhesion and humoral factors [29]. Among these is the angiopoietin-1/Tie-2 axis. HSC express the receptor tyrosine kinase Tie2 and adhere to osteoblasts that express angiopoietin-1 in the bone marrow niche. The interaction of Tie-2 with its ligand angiopoietin-1 maintains in vivo the long-term repopulating activity of HSC. Angiopoietin-1 promotes in HSC a state of quiescence and their adhesion to the bone, protecting the HSC compartment from myelosuppressive stress [30]. Adhesion of HSC to osteopontin present in endosteal bone via β1 integrin, is another adhesion mechanism that promotes the maintenance of HSC in the endosteal niche [31]. Among the humoral factors that control the fate of HSC is the chemokine, stromal-derived factor-1 (SDF-1), also called CXCL12. SDF-1 is produced by bone marrow stromal cells and osteoblasts, and binds to its receptor CXCR-4 expressed at the cell surface of BMDSC. It thus acts as a retention factor, promoting the maintenance of BMDSC in the osteoblastic niche [32]. The balance between the osteoblastic and the vascular niche as well as the mobilization of BMDSC to the peripheral circulation is a dynamic process that can be altered by various traumatic conditions such as chemotherapy, radiation therapy, bone marrow transplantation, hypoxia, or tumor invasion. This balance is modulated by multiple cytokines, chemokines, proteolytic enzymes and adhesion molecules [33]. Among the proteases involved, matrix metalloproteinase-9 (MMP-9) should be discussed in particular. MMP-9 is expressed by stromal cells and HSC in the bone marrow and its production is increased upon treatment with cytotoxic drugs like 5 fluorouracil (5FU) that also stimulates the release of BMDSC from the osteoblastic niche toward the vascular niche. MMP-9 exerts its proteolytic activity on several specific proteins that control the proliferation and fate of HSC in the bone marrow. First, it cleaves membrane-associated Kit-Ligand (mKit-L) expressed by stromal cells in the osteoblastic niche. As a result, a soluble form of Kit-L (sKit-L) is released which promotes the proliferation of HSC [34]. Second, MMP-9 degrades anchorage proteins such as osteopontin, enabling the migration of HSC from the osteoblastic to the vascular niche [35, 36]. Third, MMP-9 cleaves SDF-1 at a Ser4–Lys5 peptide bond that inactivates the protein [37]. SDF-1 itself enhances the expression of MMP-9 by BMDSC, thus promoting its own degradation [38]. The HSC mobilizing agent, granulocyte-colony stimulating factor (G-CSF), promotes HSC migration from the osteoblastic niche toward the vascular niche in a similar way by inducing neutrophil elastase degradation of SDF-1 [39]. By proteolytically degrading type IV collagen present in the basement membrane. MMP-9 also promotes the intravasation of BMSC into blood vessels. Thus, low levels of MMP-9 result in high levels of mKit-L, poor release of sKit-L and high levels of SDF-1 that retain HSC in the osteoblastic niche. In contrast, high levels of MMP-9 result in abundant release of sKit-L, inactivation of SDF-1 and migration of HSC toward the vascular niche and HSC differentiation [40] and intravasation into the peripheral blood circulation. This therefore explains the higher mortality and impaired bone marrow recovery observed in MMP-9 deficient mice post chemotherapy with 5FU [34].
Tumors Recruit Bone Marrow Derived Cells which Contribute to Carcinogenesis and Progression
BMD-HSC Contribute to Inflammation in the Primary Tumor
Tumors recruit inflammatory cells into their stroma though the expression of chemokines (Fig. 2). Most tumor cells produce chemokines of the two major groups CXC and CC. Typically, CXC chemokines are active on neutrophils and lymphocytes whereas CC chemokines target monocytes, eosinophils, dendritic cells, lymphocytes and NK cells [41]. Tumor cells and stromal cells also produce many other growth factors such as VEGF, placental derived growth factor (PlGF), a ligand that binds exclusively to VEGFR-1, transforming growth factor-β (TGFβ) and macrophage colony stimulating factor (M-CSF) that contribute to the recruitment of monocytes and macrophages and the promotion of their survival and differentiation into the tumor [42, 43].

The primary tumor actively recruits BMDC. Tumor cells release SDF-1, VEGF, PlGF, M-CSF and other cytokines that attract HSC and MSC from the bone marrow. In the tumor tissue, VEGFR-1 pos. cells give rise to CD45 pos. myelo-monocytic cells, macrophages and mast cells that contribute to an inflammatory reaction and to the formation of the perivasculature. By producing MMP-9 these cells further contribute to the solubilization of VEGF and other growth factors. VEGFR-2 pos. cells give rise to endothelial cells and contribute to the formation of a de novo vasculature. MSC are recruited by primary tumors and give rise to pericytes and contribute to the formation of distant metastasis by expressing CCL-5/RANTES that stimulates tumor cell migration
These inflammatory cells derive from VEGFR-1 positive HSC [44][6, 45]. Although it was originally considered that these cells incarnate the immune defense against developing tumors, there is accumulating evidence suggesting that whereas full activation of adaptive immune cells at the stage of tumor growth may result in eradication of malignant cells, chronic activation of innate immune cells at sites of premalignant growth may actually enhance tumor development [46]. Bone marrow-derived inflammatory cells contribute indeed to the transformation and proliferation of tumor cells, to the modulation of the immune response and to the development of tumor vascularization, and this process requires B lymphocytes [47].
Chronic inflammation has long been known as a predisposing factor in various human cancers such as breast, liver, colon, bladder, prostate, stomach, ovary and skin carcinoma [6, 41]. In addition to potentially mutagenic infectious agents that sustain chronic inflammation, inflammatory cells create a microenvironment favorable for the transformation of normal proliferating cells. Inflammatory cells, in particular phagocytic cells, generate reactive oxygen and nitrogen species that contribute to the formation of the mutagenic agent peroxynitrite [48, 49]. Macrophages express macrophage migration inhibitory factor (MIF), a potent cytokine that bypasses p53 regulatory functions and thus enhances the occurrence of oncogenic mutations [50]. Inflammatory cells also contribute to tumor cell proliferation by releasing growth factors like epidermal growth factor (EGF), and cytokines like interleukin-6 (IL-6) or tumor necrosis factor (TNF), as well as chemokines such as SDF-1 that enhance leukocyte recruitment [45]. Tumor associated macrophages express immunosuppressive molecules such as IL-10 and prostaglandin E-2 (PGE-2) that allow tumor cells to evade the immune system [45]. Macrophages also contribute to tumor progression by assisting tumor cells in intravasating [51, 52]. Mast cells contribute to the early stages of tumorigenesis by stimulating proliferation and malignant conversion but at the same time promoting a more differentiated and less aggressive phenotype [53, 54]. Mice deficient in mast cells are also less sensitive to the development of intestinal tumors when exposed to the carcinogen 1,2-dimethylhydrazine [54] and exhibit a decreased rate of tumor angiogenesis [55]. A similar function for tumor infiltrating neutrophils has been reported in another model of multistage carcinogenesis [56]. Through the expression of MMP-9, mast cells, macrophages and neutrophils play a critical role for the initiation of an angiogenic switch that supports the growth of transformed cells into tumors [57].
BMD-MSC Contribute to Metastasis
Bone marrow-derived mesenchymal stem cells (BMD-MSC) have also been recently shown to be recruited in large numbers in the stroma of developing tumors. Although the role of these cells in tumor progression is not fully understood, it has been recently suggested that they may promote metastasis in a breast carcinoma model. Dr. Weinberg’s laboratory has shown for example that tumor cells stimulate the expression of chemokine CCL5 (RANTES) in mesenchymal stem cells, which then acts in a paracrine manner to enhance tumor cell motility, invasion and the formation of distant metastasis [58].
BMDC Contribute to Tumor Vascularization
Both VEGFR-1 and VEGFR-2 BMDC participate in the development of tumor vascularization. Bone marrow-derived inflammatory VEGFR-1 positive cells are recruited to neo-angiogenic niches and support new blood vessel formation [59]. Among these cells, macrophages infiltrate hypoxic tumor areas and cooperate with tumor cells to promote angiogenesis. Hypoxia enhances the expression of several proangiogenic molecules by inducing the expression of the transcription factor hypoxia-inducible factors (HIF) in macrophages. As a result, inflammatory cells produce TGFβ, VEGF, PDGF, basic fibroblast growth factor (bFGF) and specific chemokines that stimulate tumor vascularization. These angiogenic chemokines include CXCL1 through CXCL8 with the exception of CXCL4 [45]. Several contain a Glu–Leu–Arg motif (ELR motif) at the NH2-terminus, and stimulate EC proliferation and chemotaxis by interacting with their receptor CXCR-2. The contribution of ELR-containing chemokines to tumor angiogenesis has been reported in various human cancers [60]. SDF-1 which is produced by tumor cells, macrophages and many stromal cells also promotes tumor angiogenesis by recruiting CXCR-4 positive BMDC and VEGF and bFGF induce CXCR-4 expression in EC [61, 62]. Proteases produced by VEGFR-1 HSC, and in particular MMP-9, play a critical regulatory role in tumor angiogenesis. MMP-9 produced by macrophages and mast cells releases ECM-bound VEGF, and therefore increases its bioavailability and biological activity [56, 63, 64]. MMP-9 also stimulates the recruitment of pericytes along EC [65, 66]. Experiments in many laboratories have shown that in mice either genetically lacking MMP-9, treated with an inhibitor of MMP or depleted in neutrophils, tumor development is characterized by reduced frequency, decreased tumor growth and reduced tumor vascularization [53, 56, 63–66]. Occasionally, VEGFR-1 positive monocytes and immature myeloid cells acquire an EC-like phenotype and can be identified in the tumor vascular endothelium [67, 68].
VEGFR-2 positive BMDC are also recruited into the tumor and contribute to the endothelium and the formation of a mature vascular network, a process called vasculogenesis or neo-angiogenesis [15, 59, 69, 70]. The level of contribution of vasculogenesis to tumor vascularization is however highly variable and depends on the tumor type, the organ site, the development stage and the mouse strain in animal models. Whereas in murine bone marrow transplantation models the precise quantification of these cells is possible using specific markers like EGFP, the quantification of these cells in human tumors is hampered by difficulties of marking and tracking BMDC [59]. As a result, reports on the percentage of these cells in tumors have been highly variable [71]. The bone marrow is also a reservoir of pericyte progenitor cells (PPC) [72]. Two subsets of PPC have been reported to be released by the bone marrow, platelet derived growth factor receptor β (PDGFRβ) positive, Sca-1+ and CD11b+ cells and CD45+, CD11b+ cells [73]. Although they express Sca-1, whether PDGFRβ+ cells originate from HSC or MSC in the marrow is unclear. Rajentie et al. [74] reported that a subpopulation of BMDC expressing the NG2 pericyte-specific proteoglycan, and the hematopoietic markers CD45 and CD11b could be detected in the perivascular space of tumors where they contribute to mural cells.
Finally, a few other BMDC that express neither VEFGR-1 or VEGFR-2 can contribute to tumor vascularization. Tie2 expressing CD13+ monocytes (TEM) identified in several mouse tumor models are recruited to tumor sites and promote angiogenesis in a paracrine manner after surrounding angiogenic tumor vessels. TEM home to peri-endothelial sites similar to pericytes, and can differentiate into α-SMA+ pericytes which then become negative for Tie-2 [75]. Precursors of dendritic cells (DC) termed “vascular leukocytes” because they express VE-cadherin in addition to CD45, CD11c, and MHC-II, also participate in tumor vasculogenesis. These cells migrate toward tumor vessels and contribute to the assembly of the tumor neovasculature [76].
Contribution of BMDC to Cancer Metastasis
The Bone Marrow Attracts Tumor Cells
In many cancer types, circulating tumor cells can be easily identified in the peripheral blood. Although the presence of these cells is often a predictor of poor clinical outcome, it does not necessarily mean that metastatic disease is present or will occur [77, 78]. However these cells can be actively recruited by the bone marrow. The mechanism by which the bone marrow recruits these circulating tumor cells in its microenvironment has been extensively studied (Fig. 3). Central to this process is the production of chemokines by bone marrow stromal cells that act as chemoattractants for tumor cells. Among those, SDF-1 seems again to play the major role. Many tumor cells express the SDF-1 receptor CXCR-4 and thus, like HSC precursor cells, will be attracted into the osteoblastic niche [79–82]. Animal experiments have demonstrated that the migration of CXCR-4 positive breast cancer cells toward the bone marrow can be blocked when cells are pretreated with the CXCR-4 inhibitor AMD 3100 or a blocking antibody against CXCR-4. Downregulation of CXCR-4 in breast cancer cells also inhibits their ability to colonize the bone, and CXCR-4 overexpressing tumor cells have a higher propensity to form bone marrow metastasis when injected intravenously in mice [83, 84]. SDF-1 is however not the only chemokine involved and other chemokines like the ELR-containing CXCL10 have been shown to attract tumor cells in the bone marrow [85]. When tumor cells reach the osteoblastic niche, they do not remain quiescent like HSC and significantly affect the homeostasis of the bone.

Tumor cells are recruited by the bone marrow where they alter the osteoblasts and osteoclast balance. SDF-1, which is abundantly present in the bone marrow, attracts CXCR-4 expressing tumor cells. When in the bone marrow tumor cells interact with osteoblasts and osteoclast precursor cells through the production of PTHrP, RANKL and other osteoclast activating factors. Alternatively, tumor cells stimulate MSC to make IL-6. As a result, osteoclasts are activated. Tumor cells also inhibit osteoblast activity and new bone formation by blocking the Wnt pathway via the production of Dkk1. This leads to an increase in bone degradation and release of growth factors
Tumor Cells Affect the Bone Microenvironment
The bone is a tissue primarily composed of type I collagen and minerals in the form of hydroxyapatite that is normally highly resistant to degradation. Under physiological conditions, it is nevertheless one of the most active tissues undergoing constant remodeling through a delicate balance of new bone formation by osteoblasts and bone degradation by osteoclasts. Osteoblasts are cells of mesenchymal origin that reside in the endosteal niche at the inner surface of the bone where they interact with hematopoietic progenitor cells [86]. Osteoclasts are cells derived from hematopoietic progenitor cells of the monocyte-macrophage lineage and are highly specialized and equipped to degrade the bone matrix upon activation. Under pathological conditions like osteoarthritis, osteoporosis or cancer metastasis, the remodeling of the bone is significantly increased and the balance disrupted [81]. The presence of tumor cells in the osteoblastic niche can shift bone remodeling toward an excess in bone degradation, which will create osteolytic bone metastases typically seen in breast cancer, multiple myeloma, thyroid cancer and neuroblastoma or toward an excess in bone synthesis and the formation of osteoblastic metastases typically observed in prostate cancer. In most cancer metastases, however, there is not pure osteolysis or osteogenesis but a combination of both processes [3, 4]. The disruption of bone remodeling seen in cancer metastasis is the result of a combination of humoral and adhesive interactions between tumor cells and stromal cells in the bone marrow microenvironment. A typical mechanism seen in breast cancer bone metastasis involves the secretion of parathyroid hormone-related peptide (PTHrP) by tumor cells [87]. In response to PTHrP, osteoblasts express the membrane bound receptor activator of NFkB ligand (RANKL), a member of the TNF-α family of cytokines and a key regulator of osteoclastogenesis [88]. RANKL binds to its receptor RANK, present at the cell surface of osteoclast precursor cells, and via NFkB activation stimulates the transcriptional expression of genes like integrin αvβ3, cathepsin K, MMP-9 and H+-ATPase necessary for osteoclast adhesion to the bone, the formation of an osteolytic lacuna and bone degradation [89]. The interaction between RANK and RANKL is regulated by osteoprotegerin (OPG), a member of the TNF receptor family produced by osteoblasts and bone marrow stromal cells that acts as a decoy receptor for RANKL, preventing its interaction with osteoclast-associated RANK [90]. This interaction leads to inhibition of osteoclast differentiation, maturation and activation. The degradation of the bone matrix by osteoclasts has another important consequence on the bone microenvironment that fuels the proliferation of tumor cells. Many growth factors that are immobilized in the bone matrix like TGF-β, IGF-I and II, PDGF, and bone morphogenic proteins (BMP) are released in a soluble form when the bone is degraded. These factors stimulate the proliferation of osteoblasts but also of tumor cells, creating a vicious circle [4]. Tumor cells can also directly activate osteoclasts without interacting with osteoblasts by secreting a variety of osteoclast activating factors (OAF) like IL-1, IL-6, MIP1-α, M-CSF or RANKL which will stimulate bone degradation. In other situations, tumor cells do not produce OAF but will stimulate the expression of OAF by MSC [91, 92]. This is, for example, the case in neuroblastoma and myeloma. Our laboratory has shown that in neuroblastoma, tumor cells do not make OAF but stimulate through a soluble factor, the expression of IL-6 by MSC, which then acts as a potent activator of osteoclasts [93]. Neuroblastoma cells express the IL-6 receptor and thus respond to IL-6 produced by MSC. The production of IL-6 by MSC not only stimulates osteoclasts but provides tumor cells with a growth and survival advantage. For example we have shown that neuroblastoma cells proliferate faster and are resistant to drug-induced apoptosis when cultured in the presence of MSC [94]. In myeloma, contact between myeloma cells and bone marrow stromal cells also stimulates the production of IL-6 by MSC and stromal cells [95]. Tumor cells not only stimulate osteoclasts but can also inhibit osteoblasts and osteogenesis. Osteoblast activation and maturation is stimulated by the Wnt pathway [96]. This pathway is negatively regulated by Dickkopf-1 (Dkk1), a soluble protein that when binding to Kremen and LRP-5/6, two membrane associated receptors, prevents the interaction of Frizzled-bound Wnt with LRP5/6 that is required for activation of the canonical Wnt pathway [97]. Breast cancer cells and myeloma cells express Dkk1 and expression of Dkk1 in these cells promotes osteolytic metastasis by inhibiting bone synthesis [98–100]. In other cases, tumor cells will promote osteoblast proliferation and activity. This is in particular the case of prostate cancer cells that express members of the bone morphogenic protein family that stimulate osteoblasts and osteogenesis [101]. Thus tumor cells that have been recruited into the bone marrow can dramatically modify the bone microenvironment into a fertile soil that provides them with a substantial proliferative and survival advantage. This explains why when a cancer has metastasized to the bone, it is generally rapidly progressive and resistant to chemotherapy.
BMDC Contribute to the Establishment of Distant Metastasis
Until recently, the accepted dogma in metastasis has been that distant organs are first colonized by a sub-population of tumor cells from the primary tumor and that inflammatory cells and EC are then recruited at these sites, where they contribute to angiogenesis and stimulate the proliferation of tumor cells. Recent provocative data however have suggested that the order of these events may well be reversed and that inflammatory cells may be the first ones to reach metastatic sites, forming pre-metastatic niches that attract circulating tumor cells [102]. Using irradiated mice transplanted with EGFP-expressing bone marrow cells and injected subcutaneously with B16 or Lewis Lung tumor cells, Kaplan et al. demonstrated that VEGFR-1 positive BMDC colonize the lung or liver at least a week before tumor cells reach these sites. They also demonstrated that tumor cells only establish metastasis at sites that are precolonized by VEGFR-1+ BMDC and that metastasis could be inhibited by treating the mice with an anti-VEGFR-1 antibody or by removal of VEGFR-1 positive cells from the marrow. The deposition of fibronectin at these pre-metastatic sites seems to be critical in attracting the VEGFR-1 positive cells that express the fibronectin-binding integrin α4β1 (VLA4). It is postulated (Fig. 4) that these cells could then alter the microenvironment leading to the release of chemokines attracting tumor cells like SDF-1. Soon after implantation of tumor cells at these sites, VEGFR-2+ cells are recruited from the bone marrow to promote neo-angiogenesis. Production of VEGF and PlGF by metastatic tumor cells stimulates angiogenesis and the recruitment of additional VEGFR-1+ BMDC. In support of the possibility of a similar mechanism in human cancer metastasis, the presence of VEGFR-1 positive cells in histological sections of human metastatic tumors has been demonstrated. Altogether the data suggest that circulating tumor cells need specific niches to establish themselves and form successful metastatic tumors. The bone marrow therefore not only is a source of local osteoblastic niches that promote the establishment of bone metastases but also of distant pre-metastatic niches that attract circulating tumor cells in distant organs where they find a favorable soil for metastasis [26, 103].

The bone marrow releases VEGFR-1 and 2 pos. precursor cells that promote the development of distant metastases. VEGFR-1 BMDC cells can colonize distant organs via adhesion to fibronectin forming pre-metastatic niches (1). These cells release SDF-1 that attracts CXCR-4 circulating tumor cells (2). When established in these pre-metastatic niches, tumor cells secrete VEGF, PlGF and other cytokines that attract VEGFR-2 pos. BMDC that will contribute to neo-angiogenesis (3)
Conclusion
It is now evident that the bone marrow plays a very unique microenvironmental role in carcinogenesis, tumorigenesis, angiogenesis and metastasis. Most of the experimental evidence discussed in this article suggests that it has a positive—and thus undesirable—effect on cancer progression. A major mechanism by which the bone marrow contributes to cancer progression is by the release of VEGFR-1 and VEGFR-2 precursor cells that are a source of mature inflammatory and vascular cells. The vast majority of the treatments currently used in human cancer, including high-dose cytotoxic and myeloablative chemotherapy, bone marrow transplantation and radiation therapy, cause an injury to the bone marrow that is followed by a phase of recovery. The effect of such injuries to the bone marrow microenvironment and in particular to the release of VEGFR-1 and -2 positive cells is so far poorly understood, but there is evidence that it may promote the release of these BMDC. For example, in animal models of limb injury the revascularization of the limb is accelerated if the mice receive low-dose irradiation, because irradiation of the bone marrow promotes the colonization of the injured limb by BMDC. Not surprisingly, this process is dependent on MMP-9 which increases the solubilization of Kit-L and the release of VEGF which both contribute to revascularization of the limb [104]. In mice treated with cyclophosphamide there is an increased release of EPC in the peripheral blood between 1 and 3 weeks after the administration of the cytotoxic agent. This release however is only observed if a bolus maximal tolerable dose is administered and not if low-dose metronomic doses are given [105, 106]. This thus supports the concept that high dose pulse chemotherapy may stimulate the release of EPC and ultimately tumor revascularization. In patients with cancer, studies have also shown an increase in EPC post high-dose chemotherapy [107]. However whether there is actually an increase in tumor vasculogenesis post administration of high-dose chemotherapy in human cancer has not been demonstrated yet. The data nevertheless suggest that pulses of high-dose chemotherapy may have an unanticipated and detrimental side effect by promoting the release of VEGFR-1 and -2 BMDC from the bone marrow osteoblastic niche into the peripheral blood circulation [108]. This may have several important and negative consequences on patient survival as it may not only promote inflammation and vasculogenesis in the primary tumor but also the formation of pre-metastatic niches and thus metastatic disease. Should this concept be correct, a better understanding of the mechanisms involved in the release of BMDC and how it is affected by injury to the bone marrow, will allow the design of new therapeutic protocols that could prevent the increased release of BMDC from the bone marrow niche (Fig. 5). For example, adding drugs that target VEGF, PlGF, SDF-1 or MMP-9 immediately after a pulse of high dose chemotherapy may prevent the release of VEGFR-1 and R2 positive cells from the bone marrow and their recruitment by the primary tumor or by pre-metastatic niches. A decrease in the levels of circulating EPC in patients with elevated VEGF levels in the peripheral blood (POEMS syndrome) treated with Bevasizumab (Avastin) has been recently shown [109]. MMP inhibitors have failed in clinical trials in the past but were tested in chronic administration in patients with end stage disease and with a reduction in the primary tumor as the therapeutic goal [110]. However when used for a short period of time between courses of intensive chemotherapy, they may be effective without unacceptable toxicity. Alternatively, intervening with SDF-1/CXCR-4 signaling with small inhibitors like AMD 3100 [111] may have a similar effect by preventing the colonization of the primary tumor by BMDC released into the peripheral blood post high-dose chemotherapy. There is clearly a fertile ground for investigation in this area that has the potential to significantly influence the way we administer chemotherapy to patients with cancer.

Revisiting the administration of high dose chemotherapy. a Recent observations suggest that there is an increase in the level of EPC after pulse high dose chemotherapy. This increase may favor tumor progression and the establishment of distant metastases if these cells are recruited by the primary tumor and by pre-metastatic niches. b The administration of agents blocking the recruitment of VEGFR-1 and 2 positive BMDC between courses of high dose chemotherapy may prevent a stimulation of neo-angiogenesis and the formation of pre-metastatic niche post chemotherapy
Abbreviations
- EC:
-
endothelial cells
- BMDC:
-
bone marrow-derived cells
- BMDSC:
-
bone marrow-derived stem cells
- GFP:
-
green fluorescent protein
- HSC:
-
hematopoietic stem cells
- MSC:
-
mesenchymal stem cells
- VEGF:
-
vascular endothelial cell growth factor
- VEGFR:
-
VEGF receptor
- CEC:
-
circulating EC
- EPC:
-
endothelial progenitor cells
- G-CSF:
-
granulocyte colony stimulating factor
- GM-CSF:
-
granulocyte-macrophage colony stimulating factor
- MMP:
-
matrix metalloproteinases
- SDF-1:
-
stromal derived factor-1
- IL:
-
interleukin
- TEM:
-
Ti-2 expressing monocytes
- PPC:
-
pericyte progenitor cells
References
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Roodman GD (2004) Mechanisms of bone metastasis. N Engl J Med 350:1655–1664
Guise TA, Kozlow WM, Heras-Herzig A, Padalecki SS, Yin JJ, Chirgwin JM (2005) Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer 5(Suppl):S46–S53
Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, Munshi NC, Anderson KC (2007) The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am 21:1007–1034
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420:860–867
Rafii S, Lyden D, Benezra R, Hattori K, Heissig B (2002) Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer 2:826–835
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–11093
Schatteman GC, Dunnwald M, Jiao C (2007) Biology of bone marrow-derived endothelial cell precursors. Am J Physiol Heart Circ Physiol 292:H1–18
Loges S, Fehse B, Brockmann MA, Lamszus K, Butzal M, Guckenbiehl M, Schuch G, Ergun S, Fischer U, Zander AR, Hossfeld DK, Fiedler W, Gehling UM (2004) Identification of the adult human hemangioblast. Stem Cells Dev 13:229–242
Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G (1998) A common precursor for hematopoietic and endothelial cells. Development 125:725–732
Eichmann A, Marcelle C, Breant C, Le Douarin NM (1993) Two molecules related to the VEGF receptor are expressed in early endothelial cells during avian embryonic development. Mech Dev 42:33–48
Bailey AS, Jiang S, Afentoulis M, Baumann CI, Schroeder DA, Olson SB, Wong MH, Fleming WH (2004) Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood 103:13–19
Rabbany SY, Heissig B, Hattori K, Rafii S (2003) Molecular pathways regulating mobilization of marrow-derived stem cells for tissue revascularization. Trends Mol Med 9:109–117
Bertolini F, Shaked Y, Mancuso P, Kerbel RS (2006) The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer 6:835–845
Bertolini F, Mancuso P, Kerbel RS (2005) Circulating endothelial progenitor cells. N Engl J Med 353:2613–2616
Urbich C, Dimmeler S (2004) Endothelial progenitor cells: characterization and role in vascular biology. Circ Res 95:343–353
Dennis JE, Merriam A, Awadallah A, Yoo JU, Johnstone B, Caplan AI (1999) A quadripotential mesenchymal progenitor cell isolated from the marrow of an adult mouse. J Bone Miner Res 14:700–709
Gronthos S, Zannettino AC, Hay SJ, Shi S, Graves SE, Kortesidis A, Simmons PJ (2003) Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J Cell Sci 116:1827–1835
Kemp KC, Hows J, Donaldson C (2005) Bone marrow-derived mesenchymal stem cells. Leuk Lymphoma 46:1531–1544
Le Blanc K (2006) Mesenchymal stromal cells: tissue repair and immune modulation. Cytotherapy 8:559–561
Prockop DJ (1997) Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 276:71–74
Etheridge SL, Spencer GJ, Heath DJ, Genever PG (2004) Expression profiling and functional analysis of wnt signaling mechanisms in mesenchymal stem cells. Stem Cells 22:849–860
Heissig B, Ohki Y, Sato Y, Rafii S, Werb Z, Hattori K (2005) A role for niches in hematopoietic cell development. Hematology 10:247–253
Yaniv I, Stein J, Farkas DL, Askenasy N (2006) The tale of early hematopoietic cell seeding in the bone marrow niche. Stem Cells Dev 15:4–16
Kaplan RN, Psaila B, Lyden D (2007) Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med 13:72–81
Taichman RS, Reilly MJ, Emerson SG (1996) Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 87:518–524
Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL (2004) Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103:3258–3264
Taichman RS (2005) Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood 105:2631–2639
Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T (2004) Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118:149–161
Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, Bertoncello I, Bendall LJ, Simmons PJ, Haylock DN (2005) Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106:1232–1239
Hattori K, Heissig B, Rafii S (2003) The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk Lymphoma 44:575–582
Arai F, Suda T (2007) Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci 1106:41–53
Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S (2002) Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109:625–637
Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW (2007) An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 26:6361–6371
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12:657–664
McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, Overall CM (2001) Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem 276:43503–43508
Yu X, Collin-Osdoby P, Osdoby P (2003) SDF-1 increases recruitment of osteoclast precursors by upregulation of matrix metalloproteinase-9 activity. Connect Tissue Res 44 Suppl 1:79–84
Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, Sandbank J, Zipori D, Lapidot T (2002) G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol 3:687–694
Kopp HG, Avecilla ST, Hooper AT, Rafii S (2005) The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology (Bethesda) 20:349–356
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, Hicklin DJ, Zhu Z, Bohlen P, Witte L, Hendrikx J, Hackett NR, Crystal RG, Moore MA, Werb Z, Lyden D, Rafii S (2002) Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med 8:841–849
Hattori K, Dias S, Heissig B, Hackett NR, Lyden D, Tateno M, Hicklin DJ, Zhu Z, Witte L, Crystal RG, Moore MA, Rafii S (2001) Vascular endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med 193:1005–1014
Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N (2002) VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417:954–958
Allavena P, Sica A, Solinas G, Porta C, Mantovani A (2008) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 66:1–9
de Visser KE, Coussens LM (2006) The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol 13:118–137
de Visser KE, Korets LV, Coussens LM (2005) De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7:411–423
Maeda H, Akaike T (1998) Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 63:854–865
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78
Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med 190:1375–1382
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J (2007) Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 67:2649–2656
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266
Coussens LM, Tinkle CL, Hanahan D, Werb Z (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103:481–490
Wedemeyer J, Galli SJ (2005) Decreased susceptibility of mast cell-deficient Kit(W)/Kit(W-v) mice to the development of 1, 2-dimethylhydrazine-induced intestinal tumors. Lab Invest 85:388–396
Starkey JR, Crowle PK, Taubenberger S (1988) Mast-cell-deficient W/Wv mice exhibit a decreased rate of tumor angiogenesis. Int J Cancer 42:48–52
Nozawa H, Chiu C, Hanahan D (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A 103:12493–12498
Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D (1999) Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13:1382–1397
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449:557–563
Kopp HG, Ramos CA, Rafii S (2006) Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol 13:175–181
Balkwill F, Coussens LM (2004) Cancer: an inflammatory link. Nature 431:405–406
Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OM, Anver MR, Kleinman HK, Murphy WJ, Oppenheim JJ (1999) Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: in vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol 154:1125–1135
Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S (2006) Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 12:557–567
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2:737–744
Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ (2002) Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst 94:1134–1142
Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, Shalinsky DR, Werb Z, Coussens LM, DeClerck YA (2004) Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res 64:1675–1686
Jodele S, Chantrain CF, Blavier L, Lutzko C, Crooks GM, Shimada H, Coussens LM, DeClerck YA (2005) The contribution of bone marrow-derived cells to the tumor vasculature in neuroblastoma is matrix metalloproteinase-9 dependent. Cancer Res 65:3200–3208
Urbich C, Heeschen C, Aicher A, Dernbach E, Zeiher AM, Dimmeler S (2003) Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation 108:2511–2516
Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC (2004) Expansion of myeloid immune suppressor Gr+ CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6:409–421
De Palma M, Naldini L (2006) Role of haematopoietic cells and endothelial progenitors in tumour angiogenesis. Biochim Biophys Acta 1766:159–166
Li B, Sharpe EE, Maupin AB, Teleron AA, Pyle AL, Carmeliet P, Young PP (2006) VEGF and PlGF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascularization. FASEB J 20:1495–1497
Goon PK, Lip GY, Boos CJ, Stonelake PS, Blann AD (2006) Circulating endothelial cells, endothelial progenitor cells, and endothelial microparticles in cancer. Neoplasia 8:79–88
Lamagna C, Bergers G (2006) The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol 80:677–681
Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G (2005) PDGFRbeta + perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol 7:870–879
Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P (2004) Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood 104:2084–2086
De Palma M, Venneri MA, Galli R, Sergi SL, Politi LS, Sampaolesi M, Naldini L (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8:211–226
Coukos G, Benencia F, Buckanovich RJ, Conejo-Garcia JR (2005) The role of dendritic cell precursors in tumour vasculogenesis. Br J Cancer 92:1182–1187
Muller V, Hayes DF, Pantel K (2006) Recent translational research: circulating tumor cells in breast cancer patients. Breast Cancer Res 8:110
Elshimali YI, Grody WW (2006) The clinical significance of circulating tumor cells in the peripheral blood. Diagn Mol Pathol 15:187–194
Sun YX, Wang JC, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, Pienta KJ, Taichman RS (2003) Expression of CXCR4, CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem 89:462–473
Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, Ben Baruch A (2001) A possible role for CXCR4, its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol 167:4747–4757
Kozlow W, Guise TA (2005) Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia 10:169–180
Strahm B, Durbin AD, Sexsmith E, Malkin D (2008) The CXCR4-SDF1alpha axis is a critical mediator of rhabdomyosarcoma metastatic signaling induced by bone marrow stroma. Clin Exp Metastasis 25:1–10
Zhang L, Yeger H, Das B, Irwin MS, Baruchel S (2007) Tissue microenvironment modulates CXCR4 expression and tumor metastasis in neuroblastoma. Neoplasia 9:36–46
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Goldberg-Bittman L, Sagi-Assif O, Meshel T, Nevo I, Levy-Nissenbaum O, Yron I, Witz IP, Ben Baruch A (2005) Cellular characteristics of neuroblastoma cells: regulation by the ELR-CXC chemokine CXCL10 and expression of a CXCR3-like receptor. Cytokine 29:105–117
Yin T, Li L (2006) The stem cell niches in bone. J Clin Invest 116:1195–1201
Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, Yoneda T, Mundy GR (1996) Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest 98:1544–1549
Kitazawa S, Kitazawa R (2002) RANK ligand is a prerequisite for cancer-associated osteolytic lesions. J Pathol 198:228–236
Teitelbaum SL (2000) Bone resorption by osteoclasts. Science 289:1504–1508
Kostenuik PJ, Shalhoub V (2001) Osteoprotegerin: a physiological and pharmacological inhibitor of bone resorption. Curr Pharm Des 7:613–635
Sohara Y, Shimada H, DeClerck YA (2005) Mechanisms of bone invasion and metastasis in human neuroblastoma. Cancer Lett 228:203–209
Callander NS, Roodman GD (2001) Myeloma bone disease. Semin Hematol 38:276–285
Sohara Y, Shimada H, Minkin C, Erdreich-Epstein A, Nolta JA, DeClerck YA (2005) Bone marrow mesenchymal stem cells provide an alternate pathway of osteoclast activation and bone destruction by cancer cells. Cancer Res 65:1129–1135
Ara T, Shimada H, Keshelava N, Metelitsa LS, Song LP, Groshen SG, Seeger RC, DeClerck YA (2008) IL-6 promotes the growth and survival of neuroblastoma cells. Cancer Res (in press)
Anderson KC, Kyle RA, Dalton WS, Landowski T, Shain K, Jove R, Hazlehurst L, Berenson J (2000) Multiple myeloma: new insights and therapeutic approaches. Hematology (Am Soc Hematol Educ Program) 72:147–165
Kim JB, Leucht P, Lam K, Luppen C, Ten Berge D, Nusse R, Helms JA (2007) Bone regeneration is regulated by wnt signaling. J Bone Miner Res 22:1913–1923
Niehrs C (2006) Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25:7469–7481
Voorzanger-Rousselot N, Goehrig D, Journe F, Doriath V, Body JJ, Clezardin P, Garnero P (2007) Increased Dickkopf-1 expression in breast cancer bone metastases. Br J Cancer 97:964–970
Qian J, Xie J, Hong S, Yang J, Zhang L, Han X, Wang M, Zhan F, Shaughnessy JD Jr., Epstein J, Kwak LW, Yi Q (2007) Dickkopf-1 (DKK1) is a widely expressed and potent tumor-associated antigen in multiple myeloma. Blood 110:1587–1594
Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Donofrio G, Bonomini S, Sala R, Mangoni M, Rizzoli V (2007) Production of Wnt inhibitors by myeloma cells: potential effects on canonical Wnt pathway in the bone microenvironment. Cancer Res 67:7665–7674
Feeley BT, Gamradt SC, Hsu WK, Liu N, Krenek L, Robbins P, Huard J, Lieberman JR (2005) Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res 20:2189–2199
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827
Psaila B, Kaplan RN, Port ER, Lyden D (2006) Priming the ‘soil’ for breast cancer metastasis: the pre-metastatic niche. Breast Dis 26:65–74
Heissig B, Rafii S, Akiyama H, Ohki Y, Sato Y, Rafael T, Zhu Z, Hicklin DJ, Okumura K, Ogawa H, Werb Z, Hattori K (2005) Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med 202:739–750
Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, Hicklin DJ, Chaplin D, Foster FS, Benezra R, Kerbel RS (2006) Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science 313:1785–1787
Bertolini F, Paul S, Mancuso P, Monestiroli S, Gobbi A, Shaked Y, Kerbel RS (2003) Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res 63:4342–4346
Furstenberger G, von Moos R, Lucas R, Thurlimann B, Senn HJ, Hamacher J, Boneberg EM (2006) Circulating endothelial cells and angiogenic serum factors during neoadjuvant chemotherapy of primary breast cancer. Br J Cancer 94:524–531
Shaked Y, Kerbel RS (2007) Antiangiogenic strategies on defense: on the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res 67:7055–7058
Rosti V, Massa M, Campanelli R, De Amici M, Piccolo G, Perfetti V (2007) Vascular endothelial growth factor promoted endothelial progenitor cell mobilization into the peripheral blood of a patient with POEMS syndrome. Haematologica 92:1291–1292
Coussens LM, Fingleton B, Matrisian LM (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295:2387–2392
Hatse S, Princen K, Bridger G, De Clercq E, Schols D (2002) Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett 527:255–262
Acknowledgments
Publication of this article was supported in part by NIH/NCI grants CA042919 and CA81403 to YDC and a grant from the Salus Sanguinis Foundation and the St-Luc Foundation to CFC and YDC. We thank Mrs. J. Rosenberg for typing the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chantrain, C.F., Feron, O., Marbaix, E. et al. Bone Marrow Microenvironment and Tumor Progression. Cancer Microenvironment 1, 23–35 (2008). https://doi.org/10.1007/s12307-008-0010-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-008-0010-7