
Abstract
Obesity, diabetes and their associated diseases are some of the greatest challenges that the world health care systems already face and with prospects of overburdening their capacities and funding. Due to decreased energetic expenditure and increased caloric intake, particularly in saturated fats, the number of people afflicted by said conditions is increasing by the day. Due to the failure, to this day, to effectively and ubiquity prevent and revert these diseases, the research into new compounds and therapeutic strategies is vital. In this review, we explain the most common dietary models of obesity and diabetes and the novel avenues of research we believe will be taken in the next few years in obesity and diabetes research. We primarily focus on the role of mitochondria and how the modulation of mitochondrial function and number as well as several promising therapeutic strategies involving metabolic regulators can positively affect the obese and diabetic status.
Similar content being viewed by others


Introduction
Diabetes and obesity
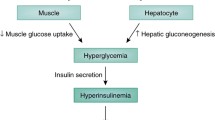
Metabolic diseases are rapidly increasing their prevalence and becoming one of the most important and costly problems affecting the world’s population and health care systems. Diabetes and its associated complications have become a worldwide epidemic, with estimates of 382 million sufferers in 2013 and 592 million people by 2,035 (International Diabetes Foundation, www.idf.org). Type 2 diabetes (T2D) is the most common form of metabolic dysfunction and is characterized by high levels of circulating glucose and overall insulin resistance. Complications from diabetes include cardiovascular diseases, optical, neuronal and renal disorders, among others (Brownlee and Cerami 1981). The incidence of T2D is typically age dependent, as more than 20 % of people older than 60 years of age have diabetes compared to only about 8 % of the population as a whole (Styskal et al. 2012).
The strongest predictor of the development of this pathology is obesity, which is rapidly increasing worldwide mainly due to an unbalanced diet and reduced physical exercise (Mann et al. 2004; Diamond 2003; Hu et al. 2001; Caterson et al. 2004). Furthermore, genetic factors also play a role in the development of obesity and T2D (Uusitupa 2005). Obesity and T2D may also share the same genetic profile; in other words, the same genetic variations may increase the risk of both obesity and T2D (Uusitupa 2005).
Insulin secretion defects and insulin resistance can also be genetically determined, whereas insulin sensitivity is highly responsive to alterations in lifestyle, which means that more physical activity, permanent weight reduction and a more balanced and adequate diet typically result in improved insulin sensitivity. Therefore, long-term obesity associated with insulin resistance and high-fat and/or sugar content diets may also, on the long run, through diverse mechanisms, result in exhaustion of β-cells, insulin resistance and cellular damage. Nevertheless, it is still unknown how much of this can be modulated by genetic factors.
Between 80 and 90 % of people with T2D are obese, so around 80 % of individuals with T2D have metabolic syndrome (Laaksonen et al. 2004). The number of obese children and adolescents is increasing, and the incidence of T2D or impaired glucose tolerance is common among young people with marked obesity (Sinha et al. 2002).
Excessive fat deposition leads to several metabolic alterations, from insulin resistance, hepatic steatosis, hyperlipidemia and cardiovascular diseases, to name a few. Obesity is responsible for the impairment of many inflammatory, endocrine and other pathways. It is of a great importance to understand how factors such as obesity can lead to diabetes and insulin resistance in order to expand therapeutic options for future treatment of these diseases. Although intensively studied, being a multipathological condition that involves several organs means that the mechanisms behind the development of these pathologies are yet to be fully understood (Cornier et al. 2008).
Molecular mechanism
The diabetic state is characterized by persistent substrate abundance in circulation. There is a stable excess of the following metabolic substrates: glucose, free fatty acids or FFA (Boden 2003), triglycerides or TG (Frohlich and Steiner 2000), lactate (Kelley and Mandarino 1990), ketone bodies (Avogaro et al. 1996) or amino acids (Vannini et al. 1982). This leads to a decrease in the activities of several metabolic pathways, since their end products are less required. As such, increased substrate (like glucose) availability leads to an increased insulin release and also to an increment in oxidative stress as, for example, a higher production of reactive oxygen species (ROS) (Hammes et al. 2003). Increased oxidizable substrates availability leads to increased mitochondrial membrane potential which, in turn, leads to diminished oxidative phosphorylation velocity and, thus, increased possibility for electrons to “jump” from the respiratory chain directly to molecular oxygen, resulting in heightened ROS generation (Rolo and Palmeira 2006). In turn, the diminished substrate utilization and increase in ATP is accompanied by an increment in NADH, in both the cytosol and in mitochondria. NAD+ and NADH values are kept relatively constant within a cell, for the ratio between them is considered a marker of metabolic status (Teodoro et al. 2013a). The increased substrate availability leads, at least in the early stages of nutrient excess exposure, to an accumulation of NADH (by both increasing the rate of NAD+ reduction and by decreasing NADH oxidation rate, as previously mentioned), leading to a decrease in metabolic activity, for example at the mitochondrial oxidative phosphorylation (OXPHOS) system and other NAD+-dependent enzymes. This reduced NAD+/NADH ratio can autofeed itself by increasing the available circulating substrates. In fact, NADH stimulates many anabolic pathways. For example, fatty acid synthesis is stimulated in the presence of excess NADH (Dahlen and Porter 1968). As mentioned before, an increase in the NADH content also causes an augmented reductive state of the mitochondrial respiratory chain, with consequent increase in ROS generation (Kukielka et al. 1994). Oxidative stress has been extensively proposed as a major cause for diabetic complications, where ROS are key players. ROS can derive from several sources, as the OXPHOS, glucose autoxidation and enzymatic activity (as NAD(P)H oxidase, lipooxygenase, cytochrome P450 monooxygenases and nitric oxide synthase (NOS)). Contributing to the increase in substrate utilization (through alterations on the insulin receptor activity), ROS augment their synthesis in an autofeedback loop (Lawlor and Alessi 2001; Vincent et al. 2002). In a normal cell, complexes I and III of the mitochondrial respiratory chain are the cell’s main ROS production sites; however, in a diabetic state, complex II overcomes these as the main ROS producer (Nishikawa et al. 2000).
Diverse studies support the hypothesis that oxidative stress promotes particular pathologies associated with diabetes. Hence, it is suggested that further investigation using antioxidant mutant mouse models could be extremely useful in the search for treatments for these pathologies related to metabolic dysfunction. For example, it has been shown that retinal damage associated with diabetes is increased in mice lacking the key antioxidant enzyme superoxide dismutase 1 (Sod1) (Dong et al. 2006), as is the rate of diabetes-induced cataract formation (Olofsson et al. 2009). Conversely, mice that overexpress Sod1 show reduced diabetes-induced retinopathy (Dong et al. 2006; Berkowitz et al. 2009). But Sod1 is not the only antioxidant enzyme studied, for mice overexpressing catalase in the kidney shows reduced renal disease associated with diabetes (Brezniceanu et al. 2008), and mice overexpressing the mitochondrial Sod2 in the heart shows reduced diabetes-induced left ventricular hypertrophy (Shen et al. 2006).
Mitochondria
Mitochondria are the powerhouses of the eukaryotic cell. Being the main ATP generation site, mitochondria play a key role in the regulation of several aspects of cell biology such as metabolism, redox status, calcium signaling and programmed cell death, to name a few (Schatz 1995). Mitochondria host, as a whole or in part, critical bioenergetic pathways, including the tricarboxylic acid (TCA) cycle, OXPHOS, the urea cycle, lipidic β-oxidation and ketogenesis.
Mitochondrial dysfunction in T2D and obesity
Mitochondrial dysfunction has been implicated in the pathogenesis of chronic metabolic diseases characterized by insulin resistance such as obesity and T2D. There is evidence that mitochondrial dysfunction may occur independently of significant changes in circulating concentrations of glucose (Boudina et al. 2005; Sloan et al. 2011), arising potentially from lipotoxicity or oxidative stress that may predate significant hyperglycemia (Sloan et al. 2011; Niemann et al. 2011). Studies have shown that insulin resistance is associated with mitochondrial dysfunction, such as reduced mitochondrial number and decreased ATP production (Kelley et al. 2002; Petersen et al. 2003). Several studies are consistent with the idea that increased caloric intake and/or obesity is associated with increased mitochondrial superoxide production. There is evidence that treatment of cell cultures with free fatty acids increases ROS production, indicating that the elevation of fatty acids in circulation may provide an additional source of excess OXPHOS substrates through increased fatty acid oxidation (Kahn and Flier 2000).
In animal models in which obesity was induced through high-nutrient feeding, levels of oxidative stress and damage are significantly increased. Levels of protein carbonylation and protein-bound 4-hydroxynonenal (a marker of lipidic peroxidation) are elevated in the adipose tissue (Sayre et al. 2006). Malondialdehyde levels (indicators of increased reaction of ROS with polyunsaturated fatty acids, PUFA) in the liver of animals fed a high-fat diet are also significantly elevated (Ling et al. 2007). These are but a few examples of all the literature available, but all these data clearly show that obesity is associated with a pro-oxidant environment and increased oxidative damage. This association is complex and multifactorial, but alterations in mitochondria function appear to play a significant role (Table 1).
Mitochondrial generation of reactive oxygen species
ROS is the collective term that generally describes a variety of molecules and free radicals derived from molecular oxygen: singlet oxygen molecules (O), superoxide anions (O2·−), hydrogen peroxide (H2O2) and hydroxyl radicals (·OH). These are products of normal metabolism, acting as “redox messengers” in intracellular signaling and regulation, allowing for communication between mitochondria and the cytosol (D’Autréaux and Toledano 2007; Mittler et al. 2011). Under normal conditions, the oxidation of glucose or fatty acids to generate ATP through mitochondrial respiration results in the production of superoxide as a by-product of the electron transport chain (ETC) (Brownlee 2001).
Mitochondria are the major sites of ROS generation in the cell, as a result of imperfectly coupled electron transport associated with OXPHOS (Boveris et al. 1976; Takeshige and Minakami 1979; Turrens et al. 1985). The outcome is a particular susceptibility of mitochondria to oxidative damage. Electrons released from the ETC incompletely reduce O2 to form O2·−, which is converted into H2O2 by manganese superoxide dismutase (MnSod, or Sod2) in the mitochondrial matrix. But what causes the imbalance in the generation of relatively low values of redox messengers to massive amounts of oxidative aggressive substances? The redox state of the respiratory chain is the primary factor for mitochondrial ROS generation (Skulachev 1996; Lambert and Brand 2004). The amplitude of the electrochemical proton gradient, which is known as respiratory control, regulates the overall rate of electron transport in the respiratory chain. When caloric intake is excessive, the electrochemical potential difference generated by the proton gradient is high, or in conditions of inhibition of the ETC complexes, the life of superoxide generating electron transport intermediates, such as ubisemiquinone, is prolonged (Skulachev 1998). This occurs because the transmembrane proton gradient and the membrane potential inherently govern the activity of the respiratory chain complexes as proton pumps. If sufficiently high, the energetic release of the electronic transport across the ETC is no longer sufficient to accumulate protons against their gradient. As such, the electronic transport slows down, giving electrons higher chances of reaching O2 and directly reduce it, generating O2·−. Increased ROS production has been shown in mitochondria isolated from skeletal muscle, kidney, liver and adipose tissue from high-fat-fed or obese animals (Anderson et al. 2009; Ruggiero et al. 2011; Raffaella et al. 2008; Curtis et al. 2010).
Obesity, diabetes, mitochondria and aging
Due to the improvements in medicine, food access and alterations in lifestyle patterns, it comes as no surprise to realize that aging-related diseases have become the most abundant cause of death in human populations, at least in the developed world (Villareal et al. 2005; Ahima 2009). Metabolic syndrome, with its associated complications (as obesity, diabetes, cardiovascular diseases, to name a few), is positively correlated with aging in humans, due to the fact that aging tissues have higher rates of ROS generation, genetic instability, and inflammatory processes. These are all been demonstrated to relate to obesity and diabetes (expertly reviewed in Hotamisligil 2006; Russel and Kahn 2007). Some of these effects have been linked to telomere shortening and oxidative damage to explain the genetic instability portion, for it has been shown that telomere shortening is accelerated by diabetes (Fuster and Andrés 2006) and that diabetes causes a galloping ROS generation increment (Valko et al. 2007). In fact, the telomere–obesity link is such an important one, and various works have reported several characteristics of said link (reviewed in Tzanetakou et al. 2012). A recent work (Minamino et al. 2009) proposed that the alterations in white adipose tissue linking obesity and aging in a genetic mice model of obesity were result of heightened activity of p53. Unsurprisingly, the same work reports that mice lacked telomerase (the enzyme responsible for maintaining telomere size) had increased insulin resistance in response to either a high-fat diet or high-sucrose diet, when compared with normal control animals in the same conditions. They also presented increased levels of inflammation and macrophage infiltration in the adipose tissue (Minamino et al. 2009). As such, there appears to be a clear link between obesity, diabetes and aging-related diseases.
But it is not just the adipose tissue to relate these phenomena, for insulin resistance in the liver (Petersen et al. 2003), skeletal muscle (Petersen and Shulman 2006), pancreas (Tschen et al. 2009) and the brain (Parrott and Greenwood 2007), to name a few, has also been related to aging-related characteristics. All these works leave an important question to be answered: Are these relationships breakable and their effects reversible? To supply and answer to these questions could revolutionize modern medicine and healthcare.
It can also be expected that mitochondrial function impairment is also heavily involved in the aging process. In fact, mitochondrial DNA (mtDNA) mutations have been related to not only diminished metabolic function, but also an accelerated aging phenotype (Larsson 2010; Lee and Wei 2012). One recent work has demonstrated that the metabolic status of the animal also causes mitochondrial alterations in aging (Gomes et al. 2013). These characteristics are also present in mitochondria from obese and diabetic individuals, in various key metabolic tissues as the liver, skeletal muscle and adipose tissue (Teodoro et al. 2008, 2013b; Patti and Corvera 2010; Gomes et al. 2012). As such, it comes as no surprise that the only known strategy to increase healthy longevity (calorie restriction) has also been shown to positively regulate mitochondrial function (Lee and Wei 2012). However, this is not without questioning, for seemingly paradoxical demonstrations of altered mitochondrial function and number appear to contribute improved health and longevity in situations of obesity and diabetes (Vernochet et al. 2012; Scialo et al. 2013). However, a careful analysis of the data demonstrates that, despite the fact that a TFAM deletion (Vernochet et al. 2012) does indeed decrease mtDNA numbers, it does not affect mitochondrial content. Also, by downplaying mitochondrial respiratory chain complex I activity, this deletion increases uncoupled respiration which, in turn, causes more substrates to be utilized (Vernochet and Kahn 2012). Surprisingly, these animals are also diet-induced obesity resistant and appear to live and healthier than their control counterparts. As such, further studies are required to elucidate these matters.
Models and diets
High-fat diets
Diet is one of the main environmental factors that contribute to the development of an obese phenotype. Along with reduced exercise, augmented caloric intake and increase in several key nutrients as high-sodium intake have lead to a dramatic elevation in the prevalence of obesity. Due to their metabolic similarity with humans, animal rodent models are useful tools for studying obesity, as they will readily increase weight when fed high-fat diets and their metabolic alterations mimic ours, for the most part.
High-fat diets (HFD) typically contain about 32–60 % of calories from fat. From a nutritional perspective, a human diet of 60 kcal % fat would be considered extreme but these are commonly used to induce rapidly induce obesity in rodents, in order to reduce experimental time courses (Ghibaudi et al. 2002; Johnston et al. 2007).
A HFD is associated with body weight increase, fat deposition throughout various organs and marked insulin resistance in animal models. Before significant increases in peripheral fat deposition occur, HFD typically increase liver fat levels as well as hepatic insulin resistance, which normally happens within days (Samuel et al. 2004).
The source of dietary fat can also modify the phenotype. Examples of these are the effect of polyunsaturated fats that increase liver fat oxidation and induce expression of proinflammatory genes (Lee et al. 2007); as examples, when compared with butter fat, olive oil reduced hepatic triglyceride accumulation and fish oil reduced liver cholesterol levels (Hussein et al. 2007).
Fatty acids can affect phenotype through a variety of mechanisms, such as gene expression, eicosanoid production and membrane receptor function. However, not all fat is equally harmful, for the health benefits of low-dose PUFA’s supplementation such as omega-3 fatty acids have been demonstrated. In animals fed similar amounts of fat, those fed diets containing fish oil (rich in these PUFAs) did not gain as much weight as those fed diets with more saturated fat (Ikemoto et al. 1996; Wang et al. 2002) and were more insulin sensitive (Buettner et al. 2006). By multiple mechanisms of action, dietary phospholipids of marine origin might thus substantially prevent and/or improve an obese phenotype and lead to the prevention and treatment strategies for obesity-associated metabolic disorders. Some works show that dietary LC n-3 PUFA administered as phospholipids from marine fish cause improved metabolic responses than triglycerides in high-fat diet-induced obese mice. Even in small doses supplementation, it increases its bioavailability, whereas more efficacious depleting of the arachnoid acid content in phospholipids augmented anti-inflammatory molecules (EPEA, DHEA) production and decreased endocannabinoids AEA and 2-AG production (Rossmeisl et al. 2012). Also, Horakova and collaborators work emphasized the idea that diet supplementation with n-3 LC-PUFA contributed to a higher efficacy of TZDs, and other insulin-sensitizing and hypolipidemic pharmaceuticals treating obesity and type 2 diabetes (Horakova et al. 2012).
High-fructose diets
An excess intake of refined carbohydrates is associated with increased weight gain, hypertriglyceridemia and insulin resistance in humans and animal models (Daly et al. 1997; Basciano et al. 2005). Purified diets containing around 60–70 % of their calories as fructose (HFrD) or sucrose are capable of elevating TG and glucose production in the liver, ultimately leading to insulin resistance and hypertriglyceridemia relative to diets containing glucose as the main carbohydrate source (Daly et al. 1997; Basciano et al. 2005). Usually, rodent chow diets contain only 4 % sucrose and less than 0.5 % of free fructose with most carbohydrate as both digestible starch and non-digestible fiber from grain sources. In contrast, low-fat purified diets can contain higher levels of sucrose, and this will depend heavily on the formula being used. It is possible to modify purified diets by manipulating the carbohydrate sources to promote metabolic syndrome while maintaining essential nutrients at recommended levels.
Fatty liver or steatosis is characterized by the accumulation of fat in the cytoplasm of hepatocytes derived from adipocytes, intestinal absorption or hepatic lipogenesis (Haque and Sanyal 2002; Browning and Horton 2004). Non-alcoholic fatty liver diseases (NAFLD) are associated with obesity and T2D and are increasing, rapidly reaching epidemic proportions.
MCD and CD diets
As with many human diseases, fatty liver in rodents is also diet inducible. A methionine and choline-deficient (MCD) diet, a choline-deficient diet (CD) or a high-fat diet (HFD) are examples. Different dietary approaches produce different severities of disease along the NAFLD spectrum and likely work by unique mechanisms.
MCD diets will rapidly induce measurable hepatic macrovesicular steatosis in rodents by 2–4 weeks and lead to inflammation and fibrosis (Sahai et al. 2004, Weltman et al. 1996). Typically, a MCD diet contains about 20 % fat by energy.
MCD diet causes weight loss in animal models (in contrast to other high-fat models) or in humans and also averting insulin resistance (Kirsch et al. 2003; Rinella and Green 2004). It also contains sucrose, which induces de novo lipogenesis and triglyceride synthesis. Pickens and collaborators showed that despite inducing the same overall level of hepatic fat accumulation, fructose was more effective than glucose at inducing hepatocellular injury in mice fed MCD diets for 21 days (Pickens et al. 2010).
A choline-deficient diet also induces fatty liver in rodents in the absence of methionine deficiency. CD diets induce steatosis, inflammation and fibrosis over 10 weeks (Teodoro et al. 2008; Fujita et al. 2010), while not leading to marked hepatitis or cirrhosis but the development of hepatocellular carcinoma.
The work conducted by Raubenheimer and co-workers used a CD diet in mice to manipulate liver fat content without affecting adipose fat stores to explore the role of liver fat accumulation in the insulin resistance and glucose intolerance of diet-induced obesity, uncovering that liver fat accumulation per se does not cause insulin resistance during high-fat feeding and that choline deficiency may shunt potentially toxic-free fatty acids toward innocuous storage triglyceride in the liver. (Raubenheimer et al. 2006).
Focusing on the importance of mitochondria, we have previously investigated the impact of fatty liver on mitochondrial function in animals fed a choline-deficient diet over time. The fat accumulation causes a severe impairment of mitochondrial function, but more importantly, it also triggers a response mechanism, designed to withstand the challenge created by the steatosis, on the bioenergetic level. This study showed that mitochondria play a central role in the development of the non-alcoholic steatosis, particularly on the oxidative phosphorylation efficiency level (Teodoro et al. 2008). Petrosillo and colleagues also demonstrated that impairment of complex I activity after 30 weeks of feeding with a choline-deficient diet is caused by ROS-dependent cardiolipin peroxidation (Petrosillo et al. 2007).
A choline-deficient l-amino acid-defined (CDAA) diet leads to the development of typical non-alcoholic steatohepatitis (NASH)-like pathology, with lobular inflammation and fibrosis in a relatively short period of time (Takeuchi-Yorimoto et al. 2013). Steatosis and inflammation improved in a rat model of CDAA (choline-deficient l-amino acid-defined) diet-induced NASH on exchange for a CSAA (choline-supplemented l-amino acid-defined) diet, but persistent fibrosis, evidences of continued oxidative stress, hypoxia, and neovascularization, were still noted (Takeuchi-Yorimoto et al. 2013).
Differences between models
The mechanisms involved with liver fat accumulation on CD diets may be different from those at work during MCD diet feeding (Kulinski et al. 2004). Distinctly from the MCD model, fatty liver induced by a CD diet is not associated with weight loss, which might make CD diets more advantageous than MCD. After 7 weeks, the MCD diet induces higher scores of liver inflammation and steatosis than the CD diet (Veteläinen et al. 2007). But the CD-fed rats gained weight, were insulin resistant and had higher plasma lipids than the MCD group animals (Veteläinen et al. 2007). When fed for equal intervals of time, HFD feeding results in significantly lower liver fat levels compared to what accumulates on an MCD diet (Romestaing et al. 2007). HFD feeding does not produce liver fibrosis and only mild steatosis as compared to MCD diets (Anstee and Goldin 2006).
The work conducted by Cong and co-workers (Cong et al. 2008) modifies a HFD (60 % of calories) to simultaneously contain low levels of methionine and choline. C57BL/6 mice were fed the diet for 23 weeks and developed obesity, insulin resistance, dyslipidemia as well as liver steatosis, inflammation and fibrosis. In C57BL/6 mice, animals consuming both HFD- and fructose-/sucrose-enriched drinking water developed hepatic fibrosis while a group consuming HFD alone did not (Kohli et al. 2010). The addition of cholesterol to a HFD increases liver fat levels, signs of liver damage and produces macro- and microvesicular steatosis compared to HFD alone, in the LDLR (−/−) mouse (Subramanian et al. 2011). In Sprague–Dawley rats, vitamin D deficiency within a HFD worsened NAFLD versus HFD alone (Roth et al. 2012).
Therapeutic strategies
Introduction
For most obese patients, the most common way to counteract obesity is a combination of one or more therapeutic strategies, based on:
-
Diet and lifestyle
-
Surgery
-
Pharmacological approach
Obviously, to switch and maintain a low-calorie healthier diet and some form of physical exercise is key to most anti-obesity treatments. Easier said than done, not just due to relapse into old habits, but also because obese people have more difficulty to perform physical exercise and, in some cases, the moderate but constant exercise required for noticeable effects can be harmful (e.g., patients with cardiac impairments, atherosclerosis). As such, the development of a “magic bullet” or therapeutic strategy that could lead to decreased obesity with minimal effort is extremely attractive, albeit probably unlikely to exist.
If exercise and diet are difficult and/or are not producing any noticeable effects, it is possible for the practitioner to prescribe a pharmacological compound to aid/drive the improvement of obesity, usually by attacking the obesity-associated T2D. Unfortunately, such compounds are usually not very effective, since they can lead to increased obesity (e.g., thiazolidinediones, TZDs) or are not very effective on the long run (e.g., insulin secretagogues). As such, it comes as no surprise that the development of cutting edge compounds and therapies could vastly increase the metabolic status of obese/diabetic patients.
In extreme cases, one effective (although risky) strategy is based on bariatric surgery or the reduction of stomach volume. Gastric bypass, among other procedures, can have a dramatic effect on body weight, but is also dependent on lifestyle changes and carries all the risks and costs of surgical procedures.
As such, the search continues for the magic bullet. Candidates are tested, either of chemical or biological origin, new or well known, individually or in combination, to try to unveil which can cause the great breakthrough in the twenty-sirst century epidemic. The new drugs must act in one of three different ways: It will reduce appetite, reduce nutrient absorption or increase the basal metabolic rate. In this review, we will explore some of the novel strategies being currently tested.
Commonly used drugs
Phentermine is a commonly used drug prescribed to obese individuals for short-term use, in combination with diet and exercise (Nelson and Gehlert 2006). It is an appetite suppresser based on the chemical structure of amphetamine, and, as such, it comes with no surprise that overuse can lead to several complications, as addiction and cardiovascular problems (Yanovski 2005). There are currently various classes of regularly prescribed drugs to treat obesity on for long-term use, most notably sibutramine and orlistat.
The first is a serotonin and noradrenaline reuptake inhibitor (also chemically similar to amphetamine, but with a distinct mechanism of action—forced secretion versus reuptake inhibition), which limits food intake by giving a sensation of satiety (Heal et al. 1998). It has been related to various cases of hypertension, cardiovascular complications and strokes, being withdrawn from the market in several countries, but it is still available in the United States.
Orlistat is a stable, potent pancreatic lipase inhibitor, with capacity to reduce obesity and prevent the onset of T2D (Torgerson et al. 2004). It has, unsurprisingly, side effects, most notably steatorrhea, lipid-soluble vitamins malabsorption and possibly liver damage. To counter this, manufacturers advise patients to reduce fat intake, which, to some extent, beats the purpose of the compound.
Lorcaserin is a serotonin receptor agonist; thus, their activation on the hypothalamus leads to proopiomelanocortin release and weight loss through satiety, a mechanism not entirely dissonant from Sibutramide and other amphetamine-like compounds (Smith et al. 2008), without the cardiotoxic side effects. Other serotonin receptor agonists such as fenfluramine and dexfenfluramine were highly effective as weight-loss drugs, but had to be removed from the market due to severe cardiac toxicity (Rothman and Baumann 2009).
Phentermine has also been used in combination with topiramate, a strategy focused on satiety as described above combined with the anorexic side effects of topiramate, an anticonvulsant (Gadde et al. 2011).
Rimonabant tackles the problematic in a different fashion. It is an antagonist of the cannabinoid receptor CB1, thus leading to food intake suppression (Fong and Heymsfield 2009). It has been since removed from the market (due to side effects such as depression), but it pointed to the study of CB1 receptors on the fight against obesity.
Metformin, a common anti-diabetic agent (inhibits hepatic gluconeogenesis and increases muscular glucose uptake) has also been shown to decrease obesity, albeit as a secondary effect (Bray and Greenway 1999). Other anti-diabetic compounds can also lead to decreased obesity, but mostly as an indirect effect.
These are some of the most known anti-obesity drugs in use or discontinued in the last 20 years. The focus was primarily on achieving satiety or decreasing absorption, with sometimes encouraging results. Wong et al. (2012) show that the most promising new compounds currently on test are strapped by the same mechanisms and ideas. Thus, the development of compounds that act in all new ways is essential.
New approaches to an old problem
To come up with new therapeutic strategies that fall out of the range of previously utilized ones, one must contemplate different targets for acting upon. The above-mentioned drugs all fall on the scope of nutrient malabsorption (typically by inhibiting pancreatic lipase) or on altered neuronal signaling into tricking the organism to think it has had a full meal. We will now briefly elaborate on some interesting metabolic modulators that have been showing promising results in basic science works.
Sirtuins and AMPK, nutrient sensors
Sirtuins are a class of NAD+-dependent deacetylases. By being dependent on the levels of oxidized NAD, sirtuin activity on gene transcription can be classified as a nutrient-sensitive action. Sirtuins activity as gene transcription modulators has been explored in various fields of investigation, from aging, to obesity, diabetes and Alzheimer’s, to name a few (Yamamoto et al. 2007). Obviously, our focus here is on its ability to modulate metabolism.
The first data on the importance of SirT1 on metabolic regulation rose from SirT1-null mice, which demonstrated decreased insulin release, while overexpression of SirT1 causes increased insulin response to glucose (Moynihan et al. 2005). Also, SirT1 deacetylates and thus activates peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1α (PGC-1α), a known master regulator of metabolism and mitochondrial biogenesis, whose activity is directly related to improved metabolic status (Nemoto et al. 2005). Paradoxically, SirT1 inhibits uncoupling protein-2 (UCP2) expression, leading to increased mitochondrial coupling and thus reducing substrate utilization (Moynihan et al. 2005). Conversely, PPARγ’s expression is downregulated by SirT1, leading to decreased adipogenesis and increased lipolysis (Picard et al. 2004). SirT1 activation of PGC-1α at brown adipocytes leads to increased mitochondrial biogenesis and thus increased thermogenic dissipation of excess nutrients (Lagouge et al. 2006). There are many other excellent works on SirT1 effects on metabolic regulation, which fall out of the scope of this review (see Yamamoto et al. 2007). These works are sometimes conflicting and make it difficult to understand what are the effects of SirT1 on metabolism. In fact, until very recently, the role of resveratrol (the most famous natural SirT1 activator) was questioned (Hubbard et al. 2013). SirT1 is also required for resveratrol beneficial effects, independent of the AMP-activated kinase (AMPK), another metabolic sensor (discussed below) (Price et al. 2012).
SirT1 is the most famous and most studied sirtuin, but is far from being the only one. Another extremely important sirtuin is the normally mitochondrial native SirT3. The expression of SirT3 in white and brown adipose tissue (WAT and BAT, respectively) is induced by calorie restriction and cold exposure, respectively. The most interesting fact about SirT3 is that its constitutive expression leads to increased levels of PGC-1α and UCP1, thus making it a very attractive target for obesity reduction (Figs. 1, 2).

Schematic representation of the convergence of pathways of the proposed therapeutic targets. The modulations of the activity of both AMPK and sirtuins (as, for example, the discussed Sirt1) lead to a potent activation of PGC-1α which, in turn, binds to an increase in the transcriptional activity of key target transcription factors. One example is PPARγ, a key transcription factor for metabolism. By acting on these targets, it is possible to elucidate molecular pathways and unveil therapeutic targets for exploration in the near future

The possibility of generating brown-like cells (“brite” adipocytes) from white adipocyte depots is an attractive therapeutic strategy, for these cells have an immense potential for energy consumption, rather than energy storage. As such, an isolated or combination therapy that can lead to the increase in “brite” cell content can cause a massive alteration of one’s metabolic status
Other effects of sirtuins exist, and other sirtuins have metabolic activity (e.g., SirT4 regulates insulin levels). However, except for resveratrol, practically no other agonist (specific or unspecific) for any sirtuin exists or is studied. As such, research unto new compounds that either mimic (with stronger activity) resveratrol or just compounds that modulate other sirtuins can lead to novel anti-obesity and anti-diabetic drugs (Milne et al. 2007). One example is berberine, a traditional Chinese medicine compound that we have shown to improve obesity and mitochondrial function (Gomes et al. 2012), with a strong involvement of SirT3 (Teodoro et al. 2013b).
AMPK is an important metabolic sensor and regulator being involved in, among other effects, glucose uptake, lipidic β-oxidation and mitochondrial biogenesis, with vast effects on several organs, ranging from the liver to brain, from WAT and BAT to skeletal muscle, i.e., all metabolically-relevant tissues (Winder 2001). Since AMP activates AMPK, a rise in this adenosine nucleotide (with concomitant decrease in ATP) signals the cell to begin energy-burning processes in order to generate ATP. As paralleled by NAD+ and SirT1, this property (and subsequent downstream effects) can be explored (and have been extensively studied) for obesity management. While SirT1 deacetylates proteins and histones, AMPK phosphorylates and alters proteins’ activity (either increasing or decreasing) (Hardie et al. 2012). AMPK has a similar effect to insulin, for it promotes GluT1 activation and Glut4 migration to the cellular membrane and thus increased glucose uptake and oxidation (Barnes et al. 2002; Pehmøller et al. 2009). AMPK also appears to mediate fatty acid uptake in cardiac cells (Habets et al. 2009). But AMPK not only improves the uptake of nutrients, it also leads to increased oxidation of said nutrients, mainly by the inhibition of acetyl-CoA carboxylase (ACC) and thus increase in mitochondrial import of fatty acids (Merrill et al. 1997) and by increasing the glycolytic rate (Marsin et al. 2002).
Another key effect of AMPK is on mitochondrial biogenesis. Unsurprisingly, AMPK phosphorylates and activates PGC-1α, thus leading to increased mitochondrial content, particularly in skeletal muscle (Winder et al. 2000). Finally, AMPK can also activate (and be activated) by SirT1, by increasing cellular NAD+ levels (Cantó et al. 2010).
As with sirtuins, these are but some effects of AMPK on metabolism (AMPK is also involved in many other biological functions—see Hardie et al. 2012). AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) is the well-known AMPK synthetic agonist, but studies on its effects on the long-term activation of AMPK (for a continuous therapeutic use) are surprisingly lacking.
Thiazolidinediones (TZDs)
TZDs (or how they are also known, glitazones) are a class of anti-diabetic agents that activate PPARγ, promoting its heterodimerization with the retinoid X receptor (RXR) and leading to gene transcription. This leads to various effects, not all beneficial, which puts the use of such compound into perspective. Nevertheless, the overall global effect is a potent insulin resistance decrease (Cariou et al. 2012). Among these effects, TZDs are known to increase insulin sensitivity, promote adiponectin release and decrease of inflammatory cytokines. Conversely, they are also known to lead to adipocyte differentiation and increased lipogenesis, as well as decreasing circulating leptin levels, thus increasing food intake. These effects are confusing at best, and given the natural agonists of PPARγ (e.g., polyunsaturated fatty acids, PUFA), that also demonstrate beneficial actions, one is tempted to think that the story behind PPARγ is still mostly untold. In WAT, PPARγ activation by TZDs leads to increased lipid uptake and triglyceride deposition (Hevener et al. 2003). On a first glance, this might seem a bad thing, but the removal of fat from circulation and, most importantly, preventing its deposition on the liver and skeletal muscle is immensely beneficial in terms of insulin resistance, in what is known as the “lipid steal hypothesis.” Also at the WAT level, an increase in adiponectin synthesis and decrease in inflammatory cytokines contribute to improved insulin sensitivity (Yu et al. 2002; Weisberg et al. 2003). TZDs also appear to improve hepatic steatosis, fibrosis and inflammation. This might seem surprising, even paradoxical, for TZDs increase hepatic fat deposition, and thus, one would believe that it would increase steatosis. In fact, WAT fat deposition stimulated by TZDs outweighs fat deposition in the liver, with the overall result being an improvement on hepatic steatosis (Belfort et al. 2006). PPARγ agonism by TZDs also improves atherosclerosis, by reduction of inflammation and circulating cholesterol (Rigamonti et al. 2008). Finally, despite the fact that there is no PPARγ expression in skeletal muscle, these global metabolic effects improve muscle metabolism and insulin sensitivity.
Despite these promising data, TZDs have, by the most part, removed from the market due to severe hepatotoxicity and suspected cardiotoxicity. Since it appears that there is a dose-dependent relationship between TZDs and their side effects, one would think that it would be a simple case of reducing the dosage, which can be a hard act to balance. Dual PPAR agonism (α and γ) is an interesting therapeutic strategy that has the benefits of PPARγ activation and PPARα stimulation of lipid oxidation; however, some compounds with dual PPAR agonism activity have already been shelved due to carcinogenesis and cardiotoxicity (Rubenstrunk et al. 2007). Finally, it is possible to modulate PPARγ activity in other ways. PPARγ is phosphorylated (causing a decrease in its insulin-sensitizing effects, but not on the adipogenic effects) by cyclin-dependent kinase 5 (CDK5). By targeting CDK5 alone or in simultaneous treatment with a PPARγ agonist might be a powerful therapeutic strategy for the future (Choi et al. 2011). As its name implies, PPARγ is also regulated by PGC-1α (Puigserver et al. 1999) and has a negative regulatory relationship with SirT1, which begs for further studies and the comprehensive understanding of all these interactions (Picard et al. 2004; Han et al. 2010).
Brown adipose tissue and “brite” cells
Brown adipose tissue, BAT, has evolved into being a primordial source of heat generation in newborns and small mammals, by presenting a high content on UCP1-expressing mitochondria (UCP1 is actually a hallmark protein of BAT) (Cannon and Nedergaard 2004). When activated, BAT generates a high metabolic rate, sustained by a rather large rate of substrate oxidation, which are obtained both from its lipid droplets and from circulation (Bartelt et al. 2011). As such, the overactivation of BAT could prove a highly effective therapeutic strategy for obesity. It was thought that BAT was not present in adult humans, voiding such therapeutic approaches. However, this has since been shown erroneous, and as such an interest in non-shivering thermogenesis as a mean to thwart obesity has boomed in recent years (Whittle 2012). However, Vosselman and collaborators quickly ended such hopes by demonstrating that, despite present, adult human BAT is hardly an effective way to reduce obesity (Vosselman et al. 2012). While one might rashly scrap this promising therapeutic approach, it should be made to notice that adipocytes are highly plastic cells. This means that, given the right stimuli, white adipocytes can be, to some extent, converted into brown-like cells, the so-called “brite” or beige adipocytes (the opposite, i.e., the conversion of brown into brite is also possible). As such, the next “big thing” in metabolic research is the conversion of white into brown adipocytes, thus leading to increased basal metabolic rates, burning more fuel and decreasing obesity. The ways by such phenomena occur, and what are the regulators and effectors fall out of the scope of this work. Instead, we decided to focus on novel ways to induce a “brite” phenotype.
Most therapeutic strategies that reduce adiposity in white adipocytes produce metabolic alterations that are common to what is described to happen in “brite” induction, i.e., the activation of PPARα and induction of lipolysis, leptin treatment and induction of mitochondrial biogenesis (Flachs et al. 2013). These changes are normally associated with UCP1 induction and non-shivering thermogenesis. Interestingly, UCP1-null mice are obesity resistant when exposed to cold, but not at thermoneutrality (Anunciado-Koza et al. 2008). To make matters worse, the work by Nedergaard and Cannon (2013) calculates that, despite the massive increase in UCP1 expression in WAT (that arises from virtually zero), the overall contribution of these newly formed “brite” cells to the body’s basal metabolic rate is neglectable at best. This is confirmed by our recent work that demonstrates that a powerful anti-obesity agent, the bile acid chenodeoxycholic acid (CDCA), is able to reduce obesity in mice in, to some extent, an UCP1-independent fashion (Teodoro et al. 2014). As such, these works suggest that some other pathways are at play here. Flachs et al. (2011) suggest that n-3 PUFA anti-obesogenic effects are not UCP1 dependent, nor are when combined with calorie restriction, but are rather caused by increased activity of the triglyceride/free fatty acid cycle (TG/FFA cycle), a so-called metabolic futile cycle, for it consumes energy while not requiring the generation of products. This cycle could be behind many anti-obesogenic effects of countless compounds, whose effects are clear, but mechanisms are not. Research unto these metabolic pathways is probably going to become a hot topic for obesity research in the near future.
Concluding remarks
Obesity, T2D and their associated conditions have become one the most pressing healthcare problems, to the point of being considered a global pandemy. As such, an efficient therapeutic approach for these diseases is a research priority. Despite all the efforts and previous strategies, this approach still eludes researchers and practitioners. We have demonstrated, in this review, the most commonly used animal dietary models for research in obesity and T2D, highlighting some of their characteristics and key points. In line with this, we also focused in past and current therapeutic strategies and explained why they have failed or are not as efficient. As such, we focused on 3 key targets for future research that we believe conveys the most promising ideas to thwart these diseases. We focused on SirT1 and AMPK, metabolic sensors and key metabolic regulators, and their interaction with PGC-1α, a known metabolic master regulator. We have also reviewed the most current knowledge on the possibility of modulating WAT into simulating brown adipocytes and, thus, induce energy expenditure.
References
Ahima RS (2009) Connecting obesity, aging and diabetes. Nat Med 15(9):996–997
Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119(3):573–581
Anstee QM, Goldin RD (2006) Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol 87(1):1–16
Anunciado-Koza R, Ukropec J, Koza RA, Kozak LP (2008) Inactivation of UCP1 and the glycerol phosphate cycle synergistically increases energy expenditure to resist diet-induced obesity. J Biol Chem 283(41):27688–27697
Avogaro A, Crepaldi C, Miola M, Maran A, Pengo V, Tiengo A, Del Prato S (1996) High blood ketone body concentration in type 2 non-insulin dependent diabetic patients. J Endocrinol Invest 19(2):99–105
Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115(Pt 11):2433–2442
Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, Eychmüller A, Gordts PL, Rinninger F, Bruegelmann K, Freund B, Nielsen P, Merkel M, Heeren J (2011) Brown adipose tissue activity controls triglyceride clearance. Nat Med 17(2):200–205
Basciano H, Federico L, Adeli K (2005) Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2(1):5
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355(22):2297–2307
Berkowitz BA, Gradianu M, Bissig D, Kern TS, Roberts R (2009) Retinal ion regulationin a mouse model of diabetic retinopathy: natural history and the effect of Cu/Zn superoxide dismutase overexpression. Invest Ophthalmol Vis Sci 50(5):2351–2358
Boden G (2003) Effects of free fatty acids (FFA) on glucose metabolism: significance for insulin resistance and type 2 diabetes. Exp Clin Endocrinol Diabetes 111(3):121–124
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112(17):2686–2695
Boveris A, Cadenas E, Stoppani AO (1976) Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J 156(2):435–444
Bray GA, Greenway FL (1999) Current and potential drugs for treatment of obesity. Endocr Rev 20(6):805–875
Brezniceanu ML, Liu F, Wei CC, Chenier I, Godin N, Zhang SL, Filep JG, Ingelfinger JR, Chan JS (2008) Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 57(2):451–459
Browning JD, Horton JD (2004) Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 114(2):147–152
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414(6865):813–820
Brownlee M, Cerami A (1981) The biochemistry of the complications of diabetes mellitus. Annu Rev Biochem 50:385–432
Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Scholmerich J, Bollheimer LC (2006) Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol 36(3):485–501
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84(1):277–359
Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab 11(3):213–219
Cariou B, Charbonnel B, Staels B (2012) Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab 23(5):205–215
Caterson ID, Hubbard V, Bray GA, Grunstein R, Hansen BC, Hong Y, Labarthe D, Seidell JC, Smith SC Jr, American Heart Association (2004) Prevention conference VII: obesity, a worldwide epidemic related to heart disease and stroke: Group III: worldwide comorbidities of obesity. Circulation 110(18):e476–e483
Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schürer SC, Vidović D, Shulman GI, Spiegelman BM, Griffin PR (2011) Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 477(7365):477–481
Cong WN, Tao RY, Tian JY, Liu GT, Ye F (2008) The establishment of a novel non-alcoholic steatohepatitis model accompanied with obesity and insulin resistance in mice. Life Sci 82:983–990
Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, Van Pelt RE, Wang H, Eckel RH (2008) The metabolic syndrome. Endocr Rev 29(7):777–822
Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Brestoff JR, Wiczer BM, Ilkayeva O, Cianflone K, Muoio DE, Arriaga EA, Bernlohr DA (2010) Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59(5):1132–1142
D’Autréaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8(10):813–824
Dahlen JV, Porter JW (1968) Studies on the synthesis of fatty acids by a beef heart mitochondrial enzyme system. Arch Biochem Biophys 127(1):207–223
Daly ME, Vale C, Walker M, Alberti KG, Mathers JC (1997) Dietary carbohydrates and insulin sensitivity: a review of the evidence and clinical implications. Am J Clin Nutr 66(5):1072–1085
Diamond J (2003) The double puzzle of diabetes. Nature 423(6940):599–602
Dong A, Shen J, Krause M, Akiyama H, Hackett SF, Lai H, Campchiaro PA (2006) Superoxide dismutase 1 protects retinal cells from oxidative damage. J Cell Physiol 208(3):516–526
Flachs P, Rossmeisl M, Kuda O, Kopecky J (2013) Stimulation of mitochondrial oxidative capacity in white fat independent of UCP1: a key to lean phenotype. Biochim Biophys Acta 1831(5):986–1003
Flachs P, Ruhl R, Hensler M, Janovská P, Zouhar P, Kus V, Macek JZ, Papp E, Kuda O, Svobodova M, Rossmeisl M, Tsenov G, Mohamed-Ali V, Kopecky J (2011) Synergistic induction of lipid catabolism and anti-inflammatory lipids in white fat of dietary obese mice in response to calorie restriction and n-3 fatty acids. Diabetologia 54:2626–2638
Fong TM, Heymsfield SB (2009) Cannabinoid-1 receptor inverse agonists: current understanding of mechanism of action and unanswered questions. Int J Obes (Lond) 33(9):947–955
Frohlich J, Steiner G (2000) Dyslipidaemia and coagulation defects of insulin resistance. Int J Clin Pract Suppl 113:14–22
Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, Inamori M, Saito S, Iwasaki T, Terauchi Y, Maeyama S, Nakajima A (2010) Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcohol Clin Exp Res 34(Suppl 1):S18–S24
Fuster JJ, Andrés V (2006) Telomere biology and cardiovascular disease. Circ Res 99(11):1167–1180
Gadde KM, Allison DB, Ryan DH, Peterson CA, Troupin B, Schwiers ML, Day WW (2011) Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 377(9774):1341–1352
Ghibaudi L, Cook J, Farley C, van Heek M, Hwa JJ (2002) Fat intake affects adiposity, comorbidity factors, and energy metabolism of sprague-dawley rats. Obes Res 10:956–963
Gomes AP, Duarte FV, Nunes P, Hubbard BP, Teodoro JS, Varela AT, Jones JG, Sinclair DA, Palmeira CM, Rolo AP (2012) Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim Biophys Acta 1822(2):185–195
Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA (2013) Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155(7):1624–1638
Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, Kiens B, Jensen TE, Richter EA, Bonen A, Glatz JF, Luiken JJ (2009) Crucial role for LKB1 to AMPKalpha2 axis in the regulation of CD36-mediated long-chain fatty acid uptake into cardiomyocytes. Biochim Biophys Acta 1791(3):212–219
Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 9(3):294–299
Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T (2010) SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 38(21):7458–7471
Haque M, Sanyal AJ (2002) The metabolic abnormalities associated with non-alcoholic fatty liver disease. Best Pract Res Clin Gastroenterol 16(5):709–731
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13(4):251–262
Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, Cheetham SC (1998) Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine. Int J Obes Relat Metab Disord 22:S18–S28
Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM, Olefsky J (2003) Muscle-specific Pparg deletion causes insulin resistance. Nat Med 9(12):1491–1497
Horakova O, Medrikova D, Schothorst EM, Bunschoten A, Flachs P, Kus V, Kuda O, Bardova K, Janovska P, Hensler M, Rossmeisl M, Wang-Sattler R, Prehn C, Adamski J, Illig T, Keijer J, Kopecky J (2012) Preservation of metabolic flexibility in skeletal muscleby a combined use of n-3 PUFA and rosiglitazone in dietary obese mice. PLoS ONE 7(8):e43764
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867
Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, E SY, Lamming DW, Pentelute BL, Schuman ER, Stevens LA, Ling AJ, Armour SM, Michan S, Zhao H, Jiang Y, Sweitzer SM, Blum CA, Disch JS, Ng PY, Howitz KT, Rolo AP, Hamuro Y, Moss J, Perni RB, Ellis JL, Vlasuk GP, Sinclair DA (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339(6124):1216–1219
Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC (2001) Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med 345(11):790–797
Hussein O, Grosovski M, Lasri E, Svalb S, Ravid U, Assy N (2007) Monounsaturated fat decreases hepatic lipid content in non-alcoholic fatty liver disease in rats. World J Gastroenterol 13(3):361–368
Ikemoto S, Takahashi M, Tsunoda N, Maruyama K, Itakura H, Ezaki O (1996) High-fat diet-induced hyperglycemia and obesity in mice: differential effects of dietary oils. Metabolism 45:1539–1546
International Diabetes Foundation. www.idf.org/diabetesatlas
Johnston SL, Souter DM, Tolkamp BJ, Gordon IJ, Illius AW, Kyriazakis I, Speakman JR (2007) Intake compensates for resting metabolic rate variation in female C57BL/6 J mice fed high-fat diets. Obesity (Silver Spring) 15:600–606
Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Kelley DE, Mandarino LJ (1990) Hyperglycemia normalizes insulin- stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J Clin Invest 86:1999–2007
Kirsch R, Clarkson V, Shephard EG, Marais DA, Jafer MA, Woodburne VE, Kirsch RE, Hall Pde L (2003) Rodent nutritional model of non-alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol 18:1272–1282
Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, Woods SC, Seeley RJ (2010) High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52:934–944
Kukielka E, Dicker E, Cederbaum AI (1994) Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys 309(2):377–386
Kulinski A, Vance DE, Vance JE (2004) A choline-deficient diet in mice inhibits neither the CDP-choline pathway for phosphatidylcholine synthesis in hepatocytes nor apolipoprotein B secretion. J Biol Chem 279:23916–23924
Laaksonen DE, Niskanen L, Lakka HM, Lakka TA, Uusitupa M (2004) Epidemiology and treatment of the metabolic syndrome. Ann Med 36:332–346
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127(6):1109–1122
Lambert AJ, Brand MD (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: ubiquinone oxidoreductase (complex I). J Biol Chem 279(38):39414–39420
Larsson NG (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79:683–706
Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114:2903–2910
Lee GS, Yan JS, Ng RK, Kakar S, Maher JJ (2007) Polyunsaturated fat in the methionine-choline-deficient diet influences hepatic inflammation but not hepatocellular injury. J Lipid Res 48:1885–1896
Lee HC, Wei YH (2012) Mitochondria and aging. Adv Exp Med Biol 942:311–327
Ling C, Poulsen P, Simonsson S, Ronn T, Holmkvist J, Almgren P, Hagert P, Nilsson E, Mabey AG, Nilsson P, Vaag A, Groop L (2007) Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest 117:3427–3435
Mann J, DeLeeuw I, Hermansen K, Karamanos B, Karlstrom B, Katsilambros N, Riccardi G, Rivellese AA, Rizkalla S, Slama G, Toeller M, Uusitupa M, Vessby B, Diabetes and Nutrition Study Group (DNSG) of the European Association (2004) Evidence-based nutritional approaches to the treatment and prevention of diabetes mellitus. Nutr Metab Cardiovasc Dis 14:373–394
Marsin AS, Bouzin C, Bertrand L, Hue L (2002) The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem 277(34):30778–30783
Merrill GF, Kurth EJ, Hardie DG, Winder WW (1997) AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273(6 Pt 1):E1107–E1112
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450(7170):712–716
Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I (2009) A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 15:1082–1087
Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, Van Breusegem F (2011) ROS signaling: the new wave? Trends Plant Sci 16(6):300–309
Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Méneur C, Permutt MA, Imai S (2005) Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2(2):105–117
Nedergaard J, Cannon B (2013) UCP1 mRNA does not produce heat. Biochim Biophys Acta 1831(5):943–949
Nelson DL, Gehlert DR (2006) Central nervous system biogenic amine targets for control of appetite and energy expenditure. Endocrine 29(1):49–60
Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 280(16):16456–16460
Niemann B, Chen Y, Teschner M, Li L, Silber RE, Rohrbach S (2011) Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J Am Coll Cardiol 57:577–585
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Olofsson EM, Marklund SL, Behndig A (2009) Enhanced diabetes-induced cataract in copper-zinc superoxide dismutase–null mice. Invest Ophthalmol Vis Sci 50:2913–2918
Patti ME, Corvera S (2010) The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 31(3):364–395
Parrott MD, Greenwood CE (2007) Dietary influences on cognitive function with ageing: from high-fat diets to healthful eating. Ann N Y Acad Sci 1114:389–397
Pehmøller C, Treebak JT, Birk JB, Chen S, Mackintosh C, Hardie DG, Richter EA, Wojtaszewski JF (2009) Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 297(3):E665–E675
Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300:1140–1142
Petersen KF, Shulman GI (2006) Ethiology of insulin resistance. Am J Med 119(5 Suppl 1):S10–S16
Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G (2007) Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 1767(10):1260–1267
Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429(6993):771–776
Pickens MK, Ogata H, Soon RK, Grenert JP, Maher JJ (2010) Dietary fructose exacerbates hepatocellular injury when incorporated into a methionine-choline-deficient diet. Liver Int 30:1229–1239
Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15(5):675–690
Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O’Malley B, Spiegelman BM (1999) Activation of PPARgamma coactivator-1 through transcription factor docking. Science 286(5443):1368–1371
Raffaella C, Francesca B, Italia F, Marina P, Giovanna L, Susanna I (2008) Alterations in hepatic mitochondrial compartment in a model of obesity and insulin resistance. Obesity (Silver Spring) 16(5):958–964
Raubenheimer PJ, Nyirenda MJ, Walker BR (2006) A choline-deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high-fat diet. Diabetes 55:2015–2020
Rigamonti E, Chinetti-Gbaguidi G, Staels B (2008) Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol 28(6):1050–1059
Rinella ME, Green RM (2004) The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 40:47–51
Romestaing C, Piquet M-A, Bedu E, Rouleau V, Dautresme M, Hourmand-Ollivier I, Filippi C, Duchamp C, Sibille B (2007) Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond) 4:4
Rolo AP, Palmeira CM (2006) Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol 212(2):167–178
Rossmeisl M, Jilkova ZM, Kuda O, Jelenik T, Medrikova D, Stankova B, Kristinsson B, Haraldsson GG, Svensen H, Stoknes I, Sjövall P, Magnusson Y, Balvers MGJ, Verhoeckx KCM, Tvrzicka E, Bryhn M, Kopecky J (2012) Metabolic effects of n-3 PUFA as phospholipids are superior to triglycerides in mice fed a high-fat diet: possible role of endocannabinoids. PLoS ONE 7(6):e38834
Roth CL, Elfers CT, Figlewicz DP, Melhorn SJ, Morton GJ, Hoofnagle A, Yeh MM, Nelson JE, Kowdley KV (2012) Vitamin D deficiency in obese rats exacerbates NAFLD and increases hepatic resistin and toll-like receptor activation. Hepatology 55(4):1103–1111
Rothman RB, Baumann MH (2009) Serotonergic drugs and valvular heart disease. Expert Opin Drug Saf 8(3):317–329
Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B (2007) Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta 1771(8):1065–1081
Ruggiero C, Ehrenshaft M, Cleland E, Stadler K (2011) High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am J Physiol Endocrinol Metab 300:1047–1058
Russel SJ, Kahn CR (2007) Endocrine regulation of ageing. Nat Rev Mol Cell Biol 8(9):681–691
Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF (2004) Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol 287(1):G264–G273
Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279(31):32345–32353
Sayre LM, Lin D, Yuan Q, Zhu X, Tang X (2006) Protein adducts generated from products of lipid oxidation: focus on HNE and ONE. Drug Metab Rev 38:651–675
Schatz G (1995) Mitochondria: beyond oxidative phosphorylation. Biochim Biophys Acta 1271(1):123–126
Scialo F, Mallikarjun V, Stefanatos R, Sanz A (2013) Regulation of lifespn by the mitochondrial electron transport chain: reactive oxygen species-dependent and reactive oxygen species-independent mechanisms. Antioxid Redox Signal 19(16):1953–1969
Shen X, Zheng S, Metreveli NS, Epstein PN (2006) Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55:798–805
Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K, Savoye M, Rieger V, Taksali S, Barbetta G, Sherwin RS, Caprio S (2002) Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med 346:802–810
Skulachev VP (1996) Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys 29(2):169–202
Skulachev VP (1998) Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta 1363(2):100–124
Sloan C, Tuinei J, Nemetz K, Frandsen J, Soto J, Wride N, Sempokuya T, Alegria L, Bugger H, Abel ED (2011) Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes 60:1424–1434
Smith BM, Smith JM, Tsai JH, Schultz JA, Gilson CA, Estrada SA, Chen RR, Park DM, Prieto EB, Gallardo CS, Sengupta D, Dosa PI, Covel JA, Ren A, Webb RR, Beeley NR, Martin M, Morgan M, Espitia S, Saldana HR, Bjenning C, Whelan KT, Grottick AJ, Menzaghi F, Thomsen WJ (2008) Discovery and structure-activity relationship of (1R)-8-chloro-2,3,4,5-tetrahydro-1-methyl-1H-3-benzazepine (Lorcaserin), a selective serotonin 5-HT2C receptor agonist for the treatment of obesity. J Med Chem 51(2):305–313
Subramanian S, Goodspeed L, Wang S, Kim J, Zeng L, Ioannou GN, Haigh WG, Yeh MM, Kowdley KV, O’Brien KD, Pennathur S, Chait A (2011) Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res 52:1626–1635
Styskal J, Van Remmen H, Richardson A, Salmon AB (2012) Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic Biol Med 52(1):46–58
Takeshige K, Minakami S (1979) NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem J 180(1):129–135
Takeuchi-Yorimoto A, Noto T, Yamada A, Miyamae Y, Oishi Y, Matsumoto M (2013) Persistent fibrosis in the liver of choline-deficient and iron-supplemented L-amino acid-defined diet-induced nonalcoholic steatohepatitis rat due to continuing oxidative stress after choline supplementation. Toxicol Appl Pharmacol 268(3):264–277
Teodoro JS, Rolo AP, Duarte FV, Simões AM, Palmeira CM (2008) Differential alterations in mitochondrial function induced by a choline-deficient diet: understanding fatty liver disease progression. Mitochondrion 8(5–6):367–376
Teodoro JS, Gomes AP, Varela AT, Duarte FV, Rolo AP, Palmeira CM (2013a) Uncovering the beginning of diabetes: the cellular redox status and oxidative stress as starting players in hyperglycemic damage. Mol Cell Biochem 376(1–2):103–110
Teodoro JS, Duarte FV, Gomes AP, Varela AT, Peixoto FM, Rolo AP, Palmeira CM (2013b) Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: a possible role for SirT3 activation. Mitochondrion 13(6):637–646
Teodoro JS, Zouhar P, Flachs P, Bardova K, Janovska P, Gomes AP, Duarte FV, Varela AT, Rolo AP, Palmeira CM, Kopecky J (2014) Enhancement of brown fat thermogenesis using chenodeoxycholic acid in mice. Int J Obes (Lond). doi:10.1038/ijo.2013.230
Torgerson JS, Hauptman J, Boldrin MN, Sjöström L (2004) XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 27(1):155–161
Turrens JF, Alexandre A, Lehninger AL (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237(2):408–414
Tschen SI, Dhawan S, Gurlo T, Bhushan A (2009) Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 58(6):1312–1320
Tzanetakou IP, Katsilambros NL, Benetos A, Mikhailidis DP, Perrea DN (2012) “Is obesity linked to ageing?” Adipose tissue and the role of telomeres. Age Res Rev 11(2):220–229
Uusitupa M (2005) Gene-diet interaction in relation to the prevention of obesity and type 2 diabetes: evidence from the Finnish Diabetes Prevention Study. Nutr Metab Cardiovasc Dis 15(3):225–233
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Vannini P, Marchesini G, Forlani G, Angiolini A, Ciavarella A, Zoli M, Pisi E (1982) Branched-chain amino acids and alanine as indices of the metabolic control in type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetic patients. Diabetologia 22:217–219
Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Radin MJ, An D, Lee KY, Ilkayeva OR, Zingaretti CM, Emanuelli B, Smyth G, Cinti S, Newgard CB, Gibson BW, Larsson NG, Kahn CR (2012) Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 16(6):765–776
Vernochet C, Kahn CR (2012) Mitochondria, obesity and aging. Aging (Albany NY) 4(12):859–860
Veteläinen R, van Vliet A, van Gulik TM (2007) Essential pathogenic and metabolic differences in steatosis induced by choline or methione-choline deficient diets in a rat model. J Gastroenterol Hepatol 22:1526–1533
Villareal DT, Apovian CM, Kushner RF, Klein S (2005) Obesity in older adults: technical review and position statement of the American Society for Nutrition and NAASO, The Obesity Society. Am J Clin Nutr 82:923–934
Vincent AM, Brownlee M, Russell JW (2002) Oxidative stress and programmed cell death in diabetic neuropathy. Ann N Y Acad Sci 959:368–383
Vosselman MJ, van der Lans AA, Brans B, Wierts R, van Baak MA, Schrauwen P, van Marken Lichtenbelt WD (2012) Systemic β-adrenergic stimulation of thermogenesis is not accompanied by brown adipose tissue activity in humans. Diabetes 61(12):3106–3113
Wang H, Storlien LH, Huang XF (2002) Effects of dietary fat types on body fatness, leptin, and ARC leptin receptor, NPY, and AgRP mRNA expression. Am J Physiol Endocrinol Metab 282:1352–1359
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112(12):1796–1808
Weltman MD, Farrell GC, Liddle C (1996) Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111:1645–1653
Whittle A (2012) Searching for ways to switch on brown fat: are we getting warmer? J Mol Endocrinol 49(2):R79–R87
Winder WW (2001) Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle. J Appl Physiol 91(3):1017–1028
Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO (2000) Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 88(6):2219–2226
Wong D, Sullivan K, Heap G (2012) The pharmaceutical market for obesity therapies. Nat Rev Drug Discov 11(9):669–670
Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin functions in health and disease. Mol Endocrinol 21(8):1745–1755
Yanovski SZ (2005) Pharmacotherapy for obesity—promise and uncertainty. N Engl J Med 353:2187–2189
Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, Olefsky JM (2002) The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes 51(10):2968–2974
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is part of Genes and Nutrition’s “Topical Collection on Ageing”.
Rights and permissions
About this article

Cite this article
Teodoro, J.S., Varela, A.T., Rolo, A.P. et al. High-fat and obesogenic diets: current and future strategies to fight obesity and diabetes. Genes Nutr 9, 406 (2014). https://doi.org/10.1007/s12263-014-0406-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12263-014-0406-6