
Summary
Chimeric antigen receptor (CAR) T cells are genetically engineered cells containing fusion proteins combining an extracellular epitope-specific binding domain, a transmembrane and signaling domains of the T cell receptor. The CD19-CAR T cell product tisagenlecleucel has been approved by the US Food and Drug Administration and the European Medicines Agency for therapy of children and young adults under 25 years with relapsed/refractory B‑cell acute lymphoblastic leukemia (ALL) due to a high overall response rate of 81% at 3 months after therapy. The rates of event-free and overall survival were 50 and 76% at 12 months. Despite the high initial response rate with CD19-CAR‑T cells in B‑ALL, relapses occur in a significant fraction of patients. Current strategies to improve CAR‑T cell efficacy focus on improved persistence of CAR‑T cells in vivo, use of multispecific CARs to overcome immune escape and new CAR designs. The approved CAR‑T cell products are from autologous T cells generated on a custom-made basis with an inherent risk of production failure. For large scale clinical applications, universal CAR‑T cells serving as “off-the-shelf” agents would be of advantage. During recent years CAR‑T cells have been frequently used for bridging to allogeneic hematopoietic stem cell transplantation (HSCT) in patients with relapsed/refractory B‑ALL since we currently are not able to distinguish those CAR‑T cell induced CRs that will persist without further therapy from those that are likely to be short-lived. CAR‑T cells are clearly of benefit for treatment following relapse after allogeneic HSCT. Future improvements in CAR‑T cell constructs may allow longer term remissions without additional HSCT.
Similar content being viewed by others

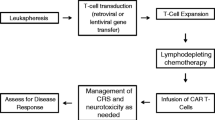
CD19-CAR‑T cells are of enormous benefit for patients with relapsed/refractory ALL.
Further improvements in CAR‑T cell constructs may allow longer term remissions without additional HSCT.
Introduction
First-line chemotherapy for patients with B‑cell acute lymphoblastic leukemia (B-ALL) younger than 65 years is intensive chemotherapy, e.g., Hoelzer protocol or hyper-CVAD (1). Allogeneic hematopoietic stem cell transplantation (HSCT) is indicated in B‑ALL for patients with a second complete remission (CR), after failure of first-line chemotherapy or upfront in first CR in B‑ALL with unfavorable prognostic factors, including complex karyotype and positive minimal residual disease (MRD) [1]. Relapsed or refractory B‑ALL is associated with a dismal prognosis with a cure rate of less than 10% [1]. CR rates with standard chemotherapy regimens are 30–40% in first relapse and 20–25% in second relapse [1]. Only 10–30% of adult patients with relapsed B‑ALL proceed to HSCT, which is the only curative therapy in this situation. Novel therapeutic options including blinatumomab and inotuzumab ozogamicin (InO) have resulted in higher response rates and longer survival (OS) than conventional chemotherapy [1]. Response to these agents enables patients to undergo HSCT as has been recently reported by Marks and colleagues where 101 of 236 patients (43%) with relapsed/refractory B‑ALL given InO, an anti-CD22 antibody conjugated to calicheamicin, proceeded to HSCT [2]. Of note, for patients with no previous HSCT who went directly to transplant after achieving remission to InO, the 2‑year survival probability was 46%. In a prospective randomized phase III study more patients with relapsed/refractory B‑ALL treated with blinatumomab, a bispecific T cell engager targeted to CD19 and CD3, compared to standard-of-care chemotherapy (SOC) achieved CR, CR with partial (CRh), or incomplete (CRi) hematologic recovery and MRD-negativity [3]. Blinatumomab prolonged OS with a median OS of 7.7 months versus 4.0 months for SOC, respectively. Patients achieving CR, CRh, or CRi after therapy with blinatumomab reportedly achieved durable responses and promising OS rates with or without subsequent HSCT [4]. Due to study limitations it is currently unclear whether patients responding to blinatumomab and obtaining MRD-negativity should undergo HSCT.
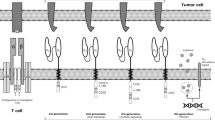
A novel cellular immunotherapeutic approach involves the genetic modification of T cells to express a chimeric antigen receptor (CAR) thereby redirecting their specificity through a mechanism independent of major histocompatibility complex (MHC) to target specific tumor antigens [5]. CARs are engineered fusion proteins combining an extracellular epitope-specific binding domain (most commonly an antibody-derived single-chain variable fragment, scFv), a hinge and transmembrane domain and signaling domains of the T cell receptor (TCR) (mostly consisting of the CD3ζ chain). Current second-generation CAR‑T constructs are combined with additional costimulatory domains such as CD28, 4‑1BB, and OX40. This enables a strong antigen-specific T cell activation without the need of TCR-MHC interactions. By their integrative capacity into the host genome, expression of CAR transgenic construct can persist independent of cell division.
Clinical results with CAR-T cells in relapsed and refractory B-ALL
Most of the targeting immunotherapies involving CAR‑T cells in B‑ALL are against the B cell surface protein CD19 and CAR‑T cell products may vary depending on the institutional design, doses, T cell activation and transduction methods. Table 1 summarizes the results of CAR‑T cell studies in patients with relapsed and refractory B‑ALL.
In a phase 2 multicenter study, 107 patients with relapsed or refractory B‑ALL were screened, 92 were enrolled and 75 (70%) received a single infusion of tisagenlecleucel, a CD19-CAR‑T cell product [6]. Among evaluable patients, the overall response rate (ORR) at 3 months was 81% with all responding patients negative for MRD as assessed by flow cytometry. In an intent-to-treat analysis of all enrolled 92 patients including subjects who discontinued study participation before tisagenlecleucel infusion, the ORR was 66%. The rates of event-free survival (EFS) and OS were 73 and 90% at 6 months and 50 and 76% at 12 months, respectively. The median duration of persistence of tisagenlecleucel in blood was 168 (range, 20–617) days and the median duration of remission was not reached at the time of reporting. Based on these results tisagenlecleucel was recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for refractory or relapsed B‑cell precursor ALL in children and young adults <25 years old.
Using quantitative polymerase chain reaction (PCR) to quantify levels of tisagenlecleucel, responding patients (n = 62/79) of two studies in pediatric B‑ALL had an approximately 2‑fold higher tisagenlecleucel expansion in peripheral blood (PB) than nonresponders with persistence measurable beyond 2 years in responding patients [7]. Clinical responses were observed across the entire dose range evaluated with no relationship between cell dose and safety. Tisagenlecleucel persistence was significantly associated with durable remission [8].
Park and colleagues reported long-term follow-up results of a single-center phase I clinical trial with CD19-CAR‑T cells in 53 adult patients with relapsed or refractory B‑ALL [9]. CR was observed in 83% of patients including 63% free of MRD. At a median follow-up of 29 (range, 1–65) months, median EFS was 6.1 months and median OS was 12.9 months, respectively. Patients with a low disease burden defined as less than 5% bone marrow (BM) blasts before therapy had longer remission duration and survival with a median EFS of 10.6 months and a median OS of 20.1 months. All 9 patients with detectable residual disease relapsed, while among 32 patients without MRD after CAR‑T cell therapy, 16 patients (50%) relapsed.
Among 30 adult patients with B‑ALL treated with a defined composition of CD4+ and CD8+ CAR‑T cells, 29 patients achieved a CR including 27 MRD− CR [10]. Gardner and colleagues administered a CAR‑T cell product of defined CD4/CD8 composition to 45 children and young adults with relapsed or refractory B‑ALL resulting in MRD− CR in 40 (89%) patients [11]. The estimated 12-month EFS was 50.8% and the estimated 12-month OS 69.5% with a median follow-up of 9.6 months. Eighteen of the 40 patients with MRD− CR experienced relapse including 7 with loss of cell-surface detection of CD19. Median time from CAR‑T cell infusion to relapse was 5.98 months.
Side-effects of CAR-T cell therapy
The potent systemic immune activation responsible for the success of CAR‑T cells also drives novel life-threatening toxicities associated with immune effector cells including cytokine release syndrome (CRS) and immune-cell associated neurotoxicity syndrome (ICANS) [12].
CRS has been defined as a disorder characterized by fever, tachypnea, headache, tachycardia, hypotension, rash, arthralgia, myalgia, and/or hypoxia by the release of proinflammatory cytokines including interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin‑6 (IL-6), and interleukin-10 (IL-10) [13]. The first symptoms occur within hours up to 14 days after CAR‑T cell therapy. Reported incidence rates of CRS vary from 40–100% with severe forms in 20–30% of patients afflicted. Disease burden has most consistently been associated with CRS severity after CAR‑T cell therapy [13]. Whether strength of T cell activation and degree of T cell expansion as well as intensity of lymphodepletion prior to CAR‑T cell infusion have an impact on development of CRS remains controversial. Since pre-existing inflammation and endothelial activation seem to be associated with severity of CRS, CAR‑T cells should not be administered to patients with pre-existing inflammation and infection [14]. Recently, the American Society of Transplantation and Cellular Therapy (ASTCT) consensus grading has been published distinguishing CRS grades according to practitioner intervention [12].
The reported incidences of grade 3 to 4 ICANS after CAR‑T cell therapy in patients with B‑ALL are between 13% [6] and 42% [15] and could be influenced by the CAR design used. The earliest symptoms of ICANS are tremor, dysgraphia, mild difficulty with expressive speech, impaired attention, apraxia, and mild lethargy [12]. Expressive aphasia, starting as impaired naming of objects, hesitant speech, and verbal perseveration, may progress to global aphasia. Within hours or days severe neurotoxicity including seizures, diffuse cerebral edema, stupor, or even coma may occur. The ASTCT consensus recently recommended use of the Immune Effector Cell-Associated Encephalopathy (ICE) score for objective grading of ICANS [12].
The pathophysiology of ICANS is poorly understood. IL‑1 triggered activation of by-standing monocytes then producing IL‑6 und leading to systemic inflammation could be an important mechanism and blockade of IL‑1 with anakinra ameliorated ICANS’ symptoms in a xenograft mouse model [16]. Santomasso and colleagues reported a significant association of severe neurotoxicity with high pretreatment disease burden, higher peak CAR‑T cell expansion, and early and higher elevations of proinflammatory cytokines in PB [15]. Patients with severe neurotoxicity had evidence of blood–cerebrospinal fluid (CSF) barrier disruption and enrichment of proinflammatory cytokines in the CSF with disproportionately high levels of IL‑6, IL‑8, MCP1, and IP10 [15].
Management of ICANS is based on corticosteroids in escalating doses depending on the severity of disease and supportive care measures [13]. Patients unresponsive to steroids could benefit from siltuximab (anti-IL‑6 chimeric monoclonal antibody), anakinra (recombinant human IL‑1 receptor antagonist) or inhibition of GM-CSF using lenzilumab [13].
Novel developments for improving CAR-T cell therapy
Despite the high initial response rate with CD19-CAR‑T cells in B‑ALL, relapses occur in a significant fraction of patients [6, 8, 10, 18]. Relapse with CD19+ leukemia cells can be the result of short in vivo persistence of CAR‑T cells, either from intrinsic deficiencies of the T cell product or an immune response to the CAR scFv [10]. Strategies to improve CAR‑T cell efficacy focus on improved persistence of CAR‑T cells by combining CD4+ CAR‑T cells and CD8+ CAR‑T cells [10, 11], enriching for central memory T cells, naive and stem memory T cells or reducing the immunogenicity of the CAR construct applying humanized CARs [18]. Efficacy of CAR‑T cell immunotherapy can be improved by use of bispecific CARs targeting CD19 and CD22 [19] or CARs with multispecificity to overcome immune escape [20]. The thymic stromal lymphopoietin receptor (TSLPR) that is overexpressed on some B‑ALL cells could be a new target for developing novel CAR constructs. Furthermore, new designs of CAR‑T cells are being explored including universal CARs that can recognize multiple targets without the need to re-engineer T cells, T cells redirected for universal cytokine-mediated killing (TRUCKs), CARs and armored CAR‑T cells modified to express cytokines, ligands, or single-chain variable fragments to elicit an enhanced antitumor immune response through turning a suppressive tumor microenvironment into a proinflammatory one. In addition, combining CAR‑T cells with checkpoint blockade therapy may reinvigorate endogenous tumor-reactive T cells that have been suppressed by the tumor microenvironment [21].
Recently, Ruella and colleagues reported a unique case of a pediatric patient with B‑ALL who relapsed with CD19-negative disease after CAR‑T cell therapy due to introduction of anti-CD19 CAR into a single leukemic B cell blast clone during CAR‑T manufacturing [22]. This case demonstrates that more stringent CAR‑T cell manufacturing methods that remove all tumor cells from the genetically engineered product are necessary, especially for protocols that involve lentiviral vectors capable of transducing nondividing cells.
Allogeneic CAR-T cells
Currently, the approved CAR‑T cell products are from autologous T cells generated on a custom-made basis by a costly and lengthy production process with an inherent risk of production failure. For large scale clinical applications universal CAR‑T cells that can serve as “off-the-shelf” ready-to-use therapeutic agents would be of advantage. Recently, gene editing technologies including TALEN (transcription activator-like effector nuclease) and CRISPR/Cas9 are being used to improve CAR‑T cells and to generate universal third party CAR‑T cells [23]. These represent a totally new generation of CAR‑T cells capable of targeting multiple antigens and/or being delivered to multiple recipients without re-editing of T cells.
Qasim and colleagues used TALENs and a lentiviral vector to generate a universal CD19-CAR‑T cell (UCART19) with disrupted expression of both CD52 and the αβ TCR for treatment of 2 pediatric patients with relapsed B‑ALL [24]. Both patients achieved a CR within 28 days of CAR‑T cell therapy and then underwent successfully a second allogeneic HSCT with minimal evidence of graft-versus-host disease (GvHD). Currently, international multicenter off-the-shelf universal UCART19 trials are ongoing for CD19+ ALL.
CAR-T cells in the context of allogeneic HSCT
The available data demonstrate that CAR‑T cells are highly active in children and adults with refractory or relapsed B‑ALL resulting in a high complete remission rate. In a subset of patients MRD-negative remissions persist with no further treatment. However, follow-up times are short and relapse after CAR‑T cell therapy has remained a challenging problem. CAR‑T cell therapy could be applied to obtain remissions for patients in first relapse who are refractory or not eligible for other therapies to achieve remission prior to allogeneic HSCT. Shalabi and colleagues reported on 52 children and young adults given CD19 and 33 patients given CD22-CAR‑T cells for treatment of relapsed/refractory B‑ALL [25]. Fifty-one patients achieved a CR including 43 with MRD-negativity and 25 subsequently underwent allogeneic HSCT. The 24-month cumulative incidences of post HSCT relapses were 13.5 and 11.3% after CD19 and CD22-CAR‑T cell therapy without an increased risk of severe GvHD. Park and colleagues observed no difference in OS between patients who completed HSCT and those who did not when comparing outcomes of the 32 patients who achieved an MRD-negative CR following CAR‑T cell infusion [9]. In the ELIANA trial which included 75 patients with relapsed or refractory B‑ALL only 8 patients (9%) underwent allogeneic HSCT in remission after CAR‑T cell therapy [6]. In view of the small patient numbers and the many differences in study design and conduct, the question whether CAR‑T cells should be administered as bridging therapy to allogeneic HSCT or are sufficiently effective by themselves cannot be answered for certain. When patients have not undergone an allogeneic HSCT previously this follow-up therapy must at least be considered.
CAR‑T cells are clearly of benefit for treatment following relapse after allogeneic HSCT since in this patient population second transplant is associated with increased toxicity and low success rates [26]. In the ELIANA study, 61% of patients had relapsed/refractory B‑ALL after allogeneic HSCT [6]. T cells were successfully collected from patients after HSCT and high remission rates without GvHD have been reported [6, 9, 17]. Thus, previous HSCT does not impact outcomes after CD19-CAR‑T cell therapy.
Conclusions
During the last few years CAR‑T cells have been frequently used for bridging to allogeneic HSCT in patients with refractory or relapsed B‑ALL [25] since we currently are not able to distinguish those CAR‑T cell induced CRs that will persist without further therapy from those that are likely to be short lived. The choice to offer HSCT to patients following CAR‑T cell therapy relies on historical experience that transplantation has been the only curative option for patients with refractory or relapsed B‑ALL. Future improvements in CAR‑T cell constructs may allow longer term remissions without additional HSCT.
Abbreviations
- A:
-
Adults
- ALL:
-
Acute lymphoblastic leukemia
- ASTCT:
-
American Society of Transplantation and Cellular Therapy
- BM:
-
Bone marrow
- BU:
-
Busulfan
- C:
-
Children
- CAR:
-
Chimeric antigen receptor
- CCR:
-
Continuous complete remission
- CD19:
-
Cluster of differentiation 19
- CR:
-
Complete remission
- CRh:
-
Complete remission with partial hematologic recovery
- CRi:
-
Complete remission with incomplete regeneration
- CRS:
-
Cytokine release syndrome
- CSF:
-
Cerebrospinal fluid
- CY:
-
Cyclophosphamide
- EFS:
-
Event-free survival
- EMA:
-
European Medicines Agency
- FDA:
-
Food and Drug Administration
- Flu:
-
Fludarabine
- GvHD:
-
Graft-versus-host disease
- HSCT:
-
Hematopoietic stem cell transplantation
- ICANS:
-
Immune-cell associated neurotoxicity syndrome
- ICE:
-
Immune effector cell-associated encephalopathy
- IFN:
-
Interferon
- IL:
-
Interleukin
- InO:
-
Inotuzumab ozogamicin
- LV:
-
Lentivirus
- MHC:
-
Major histocompatibility complex
- MRD:
-
Minimal residual disease
- NC:
-
No change
- NR:
-
No response
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PB:
-
Peripheral blood
- PCR:
-
Polymerase chain reaction
- PD:
-
Progressive disease
- PR:
-
Partial remission
- RV:
-
Retrovirus
- scFv:
-
Single-chain variable antibody fragment
- SD:
-
Stable disease
- SOC:
-
Standard of care
- TALEN:
-
Transcription activator-like effector nuclease
- TCR:
-
T cell receptor
- TNF:
-
Tumor necrosis factor
- TRUCK:
-
T cells redirected for universal cytokine-mediated killing
- TSLPR:
-
Thymic stromal lymphopoietin receptor
- UCART19:
-
Universal CD19-CAR-T cell
References
Richard-Carpentier G, Kantarjian H, Jabbour E. Recent advances in adult lymphoblastic leukemia. Curr Hematol Malig Rep. 2019;14(2):106–18.
Marks DI, Kebriaei P, Stelljes M, et al. Outcomes of allogeneic stem cell transplantation after inotuzumab ozogamicin treatment for relapsed of refractory acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2019; https://doi.org/10.1016/j.bbmt.2019.04.020.
Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Jabbour EJ, Gokbuget N, Kantarjian HM, et al. Transplantation in adults with relapsed/refractory acute lymphoblastic leukemia who are treated with blinatumomab from a phase 3 study. Cancer. 2019; https://doi.org/10.1002/cncr.32335.
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‑cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B‑cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175–84.
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59.
Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR‑T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38.
Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31.
Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38.
Borrega JG, Gödel P, Rüber MA, et al. In the eye of the storm: Immune-mediated toxicities associated with CAR‑T cell therapy. HemaSphere. 2019;3(2):e191.
Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T‑cell therapy. Blood. 2017;130(21):2295–306.
Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T‑cell therapy in patients with B‑cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958–71.
Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL‑1 and Il‑6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28.
Cao J, Cheng H, Shi M, et al. Humanized CD19-specific chimeric antigen-receptor T‑cells in 2 adults with newly diagnosed B‑cell acute lymphoblastic leukemia. Leukemia. 2019; https://doi.org/10.1038/s41375-019-0516-7.
Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B‑ALL that is naive or resistent to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20–8.
Salter AI, Pont MJ, Riddell SR. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood. 2018;131(24):2621–9.
Holzinger A, Abken H. CAR T cells: a snapshot on the growing options to design a CAR. HemaSphere. 2019;3:e172.
Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–503.
Poirot L, Philip B, Schiffer-Mannioui C, et al. Multiplex genome-edited T‑cell manufacturing platform for “Off-the Shelf” adoptive T‑cell immunotherapies. Cancer Res. 2015;75(18):3853–64.
Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B‑ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9:374.
Shalabi H, Delbrook C, Stetler-Stevenson M, et al. Chimeric antigen receptor T‑cell therapy can render patients with ALL into PCR-negative remission and can be an effective bridge to transplant. In: BMT Tandem Scientific Meeting Salt Lake City. 2018.
Lund TC, Ahn KW, Tecca HR, et al. Outcomes after second hematopoietic cell transplantation in children and young adults with relapsed acute leukemia. Biol Blood Marrow Transplant. 2019;25(2):301–6.
Yu WL, Hua ZC. Chimeric antigen receptor T‑cell (CAR T) therapy for hematologic and solid malignancies: efficacy and safety—A systematic review with meta-analysis. Cancers. 2019;11(1):47.
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Park JH, Riviere I, Wang XY, et al. Implications of minimal residual disease negative complete remission (MRD-CR) and allogeneic stem cell transplant on safety and clinical outcome of CD19-targeted 19-28z CAR modified T cells in adult patients with relapsed, refractory B‑cell ALL. Blood. 2015;126:682–6.
Callahan C, Baniewicz D, Ely B. CAR‑T cell therapy: pediatrics patients with relapsed and refractory acute lymphoblastic leukemia. Clin J Oncol Nurs. 2017;21:22–8.
Wei G, Hu Y, Pu C, et al. CD19 targeted CAR‑T therapy versus chemotherapy in re-induction treatment of refractory/relapsed acute lymphoblastic leukemia. Ann Hematol. 2018;97(5):781–9.
Weng J, Lai P, Qin L, et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J Hematol Oncol. 2018;11(1):25.
Funding
Open access funding provided by Medical University of Graz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
H.T. Greinix received honoraria as a speaker in scientific meetings from the companies Novartis and Celgene.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Greinix, H.T. Role of CAR-T cell therapy in B-cell acute lymphoblastic leukemia. memo 13, 36–42 (2020). https://doi.org/10.1007/s12254-019-00541-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-019-00541-8