
Abstract
Long noncoding RNAs (lncRNAs) participate in regulating many biological processes. However, their roles in influenza A virus (IAV) pathogenicity are largely unknown. Here, we analyzed the expression profiles of lncRNAs and mRNAs in H3N2-infected cells and mock-infected cells by high-throughput sequencing. The results showed that 6129 lncRNAs and 50,031 mRNA transcripts in A549 cells displayed differential expression after H3N2 infection compared with mock infection. Among the differentially expressed lncRNAs, 4963 were upregulated, and 1166 were downregulated. Functional annotation and enrichment analysis using gene ontology and Kyoto Encyclopedia of Genes and Genomes databases (KEGG) suggested that target genes of the differentially expressed lncRNAs were enriched in some biological processes, such as cellular metabolism and autophagy. The up- or downregulated lncRNAs were selected and further verified by quantitative real-time polymerase chain reaction (RT-qPCR) and reverse transcription PCR (RT-PCR). To the best of our knowledge, this is the first report of a comparative expression analysis of lncRNAs in A549 cells infected with H3N2. Our results support the need for further analyses of the functions of differentially expressed lncRNAs during H3N2 infection.
Similar content being viewed by others

Introduction
Swine influenza A virus (SIV) H1N1 caused the 1918 human influenza pandemic, resulting in approximately 50 million deaths (Lemon and Mahmoud 2005). Additionally, Hong Kong H3N2 flu killed one million people in 1968–1969 (Simonsen et al.1998; Dangi and Jain 2012). From April 16 to April 29, 2018, 1252 (45.4%) specimens were identified by the World Health Organization Global Influenza Surveillance and Response System laboratories as influenza A (H3N2) (http://www.who.int). Moreover, in recent years, SIV has attracted much attention owing to its ability to infect humans. Pigs have surface receptors for both avian and human influenza viruses (Ito et al.1998), and influenza viruses from humans and birds can recombine in pigs (Webster et al.1992). Among the various subtypes of SIV, H3N2 and H1N1 strains exist in human populations and cause relatively high morbidity and mortality rates (Horimoto and Kawaoka 2005). Additionally, compared with H1N1, H3N2 infection is associated with human seasonal influenza with a higher epidemic severity index, and its frequency is increasing (Simonsen et al.1997; Thompson et al.2003). Although the pathogenesis of the H3N2 subtype has been extensively studied, the underlying molecular mechanisms mediating abnormal host responses are largely unknown.
Traditional studies of host transcriptional responses to pathogen infection have mostly focused on protein-coding genes. We previously reported mitochondrial proteomic analysis of human pulmonary parenchymal cells (A549 cells) infected with swine influenza virus strain H3N2 (Wu et al.2013). We found altered expression of 24 host cell proteins (13 upregulated and 11 downregulated proteins) by two-dimensional gel electrophoresis; however, this approach was limited, and further studies are still needed to elucidate the related mechanisms.
Most mammalian genomes are transcribed into noncoding RNAs (ncRNAs), including small ncRNAs (< 200 bp) and long ncRNAs (lncRNAs; > 200 bp) (Costa 2010). With the development of high-throughput sequencing, genetic manipulation, and other biotechnologies, many functions of lncRNAs have been discovered. Studies have shown that lncRNAs play important roles in gene transcription, splicing, protein translation, protein localization, stem cell pluripotency, cell structural integrity, heat shock response, and human diseases (Szczesniak and Makalowska 2016). Moreover, lncRNAs have also been shown to have roles in innate immune responses (Carpenter et al.2013; Ilott et al.2014; Carpenter 2016; Jiang et al.2018) and host–pathogen interactions (Barriocanal et al.2014; Ouyang et al.2014; Nishitsuji et al.2016; Qiu et al.2018; Tong et al.2019). Although thousands of lncRNAs have been identified in mammalian genomes, only a few are associated with innate immunity against viral pathogenesis and influenza viruses. Importantly, lncRNAs induced by influenza virus affect several host biological processes and can also regulate infection. In addition, lncRNAs influence the feedback mode of viral infection by mediating metabolism (Wang et al.2017). However, our understanding of the specific functions of lncRNAs in influenza A virus (IAV) infection is still limited.
Therefore, to explore the function of lncRNAs in cells upon H3N2 infection, lncRNA expression profiles were analyzed in A549 cells infected with mock or H3N2 by high-throughput sequencing technology. Our data provide important insights into the interactions between host lncRNAs and H3N2 infection.
Materials and Methods
Cell Culture, Virus Infection and Preparation of Sequencing Samples
Influenza strain A/swine/Zhejiang/04/H3N2 was stored in our laboratory. A549 cells were cultured in 100-mm cell bottles, grown to approximately 80%–90% confluence, and infected with H3N2 at a multiplicity of infection (MOI) of 10. After 2 h of incubation, the culture medium was removed, and infected cells were maintained in fresh medium containing 2% fetal bovine serum. The viral titer was measured by indirect immunofluorescence assay (IFA) using monoclonal antibodies against IAV M2 protein (Feng et al.2018) at 12, 18, 24, and 36 h post-infection (hpi). The 50% tissue culture infectious dose (TCID50) was determined by the Reed-Muench method (Xue et al.2016). Uninfected A549 cells were used as a control. H3N2-infected and mock-infected cells were harvested at 24 hpi and cryopreserved at − 80 °C for subsequent total RNA extraction. All samples were analyzed in duplicate. All virus experiments were carried out in a Biosafety Level 2 laboratory.
RNA Isolation
The total RNA was extracted using TRIzol reagent (Vazyme Biotech Company, China) according to the manufacturer’s instructions. The concentration of total RNA was determined using a Nanodrop instrument (Thermo Fisher Scientific). RNA quality was assessed by determining the A260/A280 ratio, with a value of 1.8–2.0 indicating high quality. Ribo-zero-magnetic-kit (Epicentre, USA) was used to remove ribosomal RNA from the samples. RNA libraries were prepared using TruSeq RNA LT Sample Prep Kit v2 (Illumina, San Diego, CA, USA). Library sequencing was performed on an Illumina Hiseq 3000 platform (Illumina) by ShangHai Genergy Biotech (Shanghai, China). The RNA-sequencing data were deposited in the Gene Expression Omnibus database (accession number GSE135482).
Bioinformatic Analyses
Adaptors, low-quality reads, and poly-N were removed using Trim Galore software (http://www.bioinformatics.babraham.ac.uk/projects/). Quality control analysis was performed on clean data using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The filtered reads were then mapped to the human reference genome (version: Homo_sapiens.GRCh38) using STAR software suite (Dobin et al.2013). The transcripts were assembled with the mapped reads using StringTie (Pertea et al.2015). The assembled transcripts from unknown intergenic transcripts (U), a transfrag falling entirely within a reference intron (I), exonic overlap with reference on the opposite strand (X), and an intron of the transfrag overlaps a reference intron on the opposite strand were identified as novel lncRNAs. The coding ability of lncRNAs was predicted using four tools, including a predictor of lncRNAs and mRNAs based on k-mer scheme (PLEK) (Li et al.2014), Coding-Non-Coding-Index (CNCI) (Sun et al.2013), Coding Potential Calculator (CPC) (Kong et al.2007), and Coding Potential Assessment Tool (CPAT) (Wang et al.2013). The differentially expressed lncRNAs in H3N2-infected and non-infected groups were analyzed using DESeq2 (Love et al.2014).
Target Prediction of lncRNAs and Functional Analyses
Cis- and trans- analyses were implemented to predict the target genes of differentially expressed lncRNAs. The known protein-encoding genes located within a 100-kb window upstream or downstream of lncRNAs were identified as cis target genes. Co-expressed coding genes were classified as trans-regulated target genes of differentially expressed lncRNAs. GO enrichment analysis of lncRNA target genes was performed using the TopGO package (https://www.bioconductor.org/packages/release/bioc/html/topGO.html). The associated pathways of cis- or trans-regulatory target genes of the dysregulated lncRNAs were predicted by Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The statistical criteria in the analysis were as follows: P < 0.05 and false discovery rate (FDR) < 0.05. GO enrichment analysis of dysregulated mRNAs was performed using the TopGO package. The associated pathways of the dysregulated mRNAs were predicted by KEGG database. The statistical criteria in the analysis were as follows: P < 0.05 and FDR < 0.05.
Real-Time Quantitative (qPCR) Analysis of lncRNAs
In order to validate the results of high-throughput sequencing, RT-qPCR analysis was used to identify differentially expressed lncRNAs. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as an internal standard. The primer sequences are presented in Supplementary Table S1. RT-qPCR was carried out using AceQ qPCR SYBR Green Master Mix (Vazyme, China) according to the manufacturer’s instructions. The relative expression level of each lncRNA was calculated by the 2−ΔΔct method. All samples were analyzed in triplicate. Statistical analyses were performed using Student’s t tests.
Results
Replication Kinetics of H3N2 Virus in A549 Cells
To determine the propagation kinetics of H3N2 in A549 cells, we measured the viral titers at various time points after infection. Cells were infected with H3N2 at an MOI of 10 and then monitored by IFA at 12, 18, 24, and 36 hpi. The viral titer gradually increased at 12, 18, and 24 hpi and reached a maximum (105.7 TCID50/mL) at 24 hpi (Supplementary Fig. S1A and S1B). Based on these results, we selected cells infected with an MOI of 10 after 24 hpi for transcriptome analysis.
RNA-seq and Identification of Differentially Expressed lncRNAs
High-throughput RNA-seq was performed to determine the expression levels of lncRNAs in A549 cells infected with H3N2 or uninfected (mock). More than 258 billion raw base reads were generated for each sample using an Illumina Hiseq platform. After removing adaptor and low-quality sequences, average size of each clean read was 135 nt, and the clean data Q30 was above 93.45% (Supplementary Table S2).
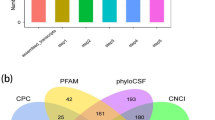
Based on various criteria, including the specific location in genome, multiple exons, length greater than 200 nt, and noncoding characteristics, transcripts were filtered by three steps to identify the annotated and novel lncRNAs. In total, 3031 transcripts were identified as novel lncRNAs (Fig. 1A).

Identification of novel lncRNAs in H3N2-infected or non-infected cells. A LncRNA screening statistics in H3N2-infected or non-infected groups. B Evaluating the coding potential using four tools. C Classification of lncRNAs based on genomic location.
Next, protein-coding or noncoding transcripts were classified using four tools, i.e., CPAT, PLEK, CNCI, and CPC (Fig. 1B). Additionally, according to the corresponding genomic locations of transcripts of known protein-coding genes, newly identified lncRNAs were categorized into four groups, i.e., intronic lncRNAs (82%), intergenic lncRNAs (12%), antisense lncRNAs (3%), and bidirectional lncRNAs (3%; Fig. 1C).
Hierarchical clustering was performed to analyze the lncRNA expression profiles in H3N2-infected or non-infected cells. Obviously, expression levels of lncRNAs were significantly altered after H3N2 infection (Fig. 2A). In total, 6129 lncRNAs were differentially expressed, including 4963 upregulated lncRNAs and 1166 downregulated lncRNAs (fold change [FC] ≥ 2, P ≤ 0.05; Fig. 2B) (Supplementary Table S3). Of the differentially regulated lncRNAs, 22 newly identified lncRNAs were altered, with log2(FC) values of more than 12, compared with the non-infected group. The lncRNA showing the greatest upregulation was MSTRG.18254.3, with a log2(FC) of more than 16; in contrast, the lncRNA showing the greatest downregulation was MSTRG.11930.8, with a log2(FC) of more than 14 (Table 1).

Characteristics of lncRNA expression levels between H3N2-infected and non-infected groups. A Differentially expressed lncRNAs were analyzed by hierarchical clustering. B Volcano plot displaying differentially expressed lncRNAs in the two groups. C Distribution of differentially expressed lncRNAs in each chromosome.
The differentially expressed lncRNAs were widely distributed in all chromosomes, although the numbers varied in different chromosomes. Most altered lncRNAs were located on chromosome 1, whereas few altered lncRNAs were located on chromosome Y (Fig. 2C).
Differential Expression of mRNAs in A549 Cells between Mock- and H3N2-infected Groups
We also detected mRNA expression levels of A549 cells after H3N2 infection. Hierarchical clustering showed that the mRNA expression profiles were significantly altered in the H3N2-infected group compared with that in the non-infected group (Fig. 3A). In total, 50,031 mRNA transcripts were found to be differentially expressed in A549 cells infected with H3N2 (FC ≥ 2, P ≤ 0.05) (Supplementary Table S4). Of the differentially expressed mRNA transcripts, 20,907 were upregulated, and 29,124 were downregulated (Fig. 3B). Moreover, 833 genes were upregulated with a FC of more than 100 after infection. In our study, the gene showing the greatest upregulation was RPL4, with a log2(FC) of 12.3, whereas the gene showing the greatest downregulation was EGFR, with a log2(FC) of −14.2 (Table 2).

Characteristics of mRNA expression levels between the H3N2-infected and non-infected groups. A Differentially expressed mRNAs were analyzed by hierarchical clustering. B Volcano plot displaying differentially expressed mRNAs in the two groups. C Distribution of differentially expressed mRNAs in each chromosome.
Similar to the distribution pattern of lncRNAs, the differentially expressed genes in H3N2-infected A549 cells were unevenly distributed among chromosomes. Most altered mRNAs were found on chromosome 1, whereas few altered mRNAs were found on chromosome Y (Fig. 3C).
Genomic Features of lncRNAs and mRNAs in A549 Cells
Next, we systematically analyzed the basic features of the lncRNAs and compared them with protein-coding genes. The lengths of lncRNA transcripts were longer than those of mRNAs (Fig. 4A). Additionally, the number of exons of lncRNAs was lower than that of mRNAs (Fig. 4B).

Genomic features of lncRNAs and mRNAs in H3N2-infected cells. A Length distribution of lncRNAs and mRNAs. B Comparison of exon numbers between lncRNA and mRNAs.
Functional Prediction of H3N2-induced lncRNAs
To better understand the roles of differentially expressed lncRNAs in H3N2-infected cells, GO and KEGG pathway analyses were used to explore the functions of cis- and trans- target genes of H3N2-induced lncRNAs. The results showed that the target genes of these lncRNAs were significantly enriched in biological processes, such as cellular metabolism. The top 20 significant GO terms are listed in Fig. 5A, 5B. The target genes of these differentially expressed lncRNAs participated in various signaling pathways, such as the B cell receptor signaling pathway and autophagy. The top 20 statistically significant enriched KEGG pathways are shown in Fig. 5C, 5D. These findings suggested that the induced lncRNAs regulated cellular metabolic processes, immunity, and autophagy during H3N2 infection. Because many genes were enriched in the cellular metabolic pathway, we further selected 10 of the most dysregulated lncRNAs to generate the cis- or trans-regulatory networks (shown in Fig. 5E, 5F).

GO enrichment and KEGG pathway analyses of target genes of H3N2-induced lncRNAs. A Top 20 significant GO biological process terms by cis-regulation. B Top 20 significant GO biological process terms by trans-regulation. C Top 20 significantly enriched pathways terms by cis-regulation. D Top 20 significantly enriched pathways terms by trans-regulation. ECis-regulatory network of lncRNAs-mRNAs, Ftrans-regulatory network of lncRNAs/mRNAs. LncRNAs are represented as triangles, and mRNAs are represented as circles. Red nodes indicate upregulation, blue nodes indicate downregulation, and color shade indicates different degrees of dysregulation.
GO and KEGG Pathway Analyses of the Dysregulated mRNAs Induced by H3N2
Dysregulated mRNAs in H3N2-infected cells were used for GO enrichment and KEGG pathway analyses. As shown in Fig. 6A, the majority of the dysregulated mRNAs were significantly enriched in some biological processes, such as cellular metabolic process, organic cyclic compound metabolic process, and cellular macromolecule metabolic process, suggesting that H3N2 infection had a profound effect on cellular metabolism in A549 cells. Furthermore, KEGG functional analyses showed that dysregulated mRNAs were significantly enriched in some pathways (Fig. 6B), including citrate cycle (tricarboxylic acid [TCA] cycle), DNA replication, biosynthesis of unsaturated fatty acids, and autophagy. Taken together, these results suggested that the differentially expressed mRNAs belonged to multiple pathways.

A Top 20 significant GO biological processes of differentially expressed mRNAs. B Top 20 significantly enriched pathways terms of differentially expressed mRNAs.
Validation of Differentially Expressed lncRNAs by RT-qPCR
RT-qPCR was performed to further detect differentially expressed lncRNAs in sequencing data. Among the 6129 differentially expressed lncRNAs, we validated seven upregulated and two downregulated lncRNAs by RT-qPCR (Fig. 7A) and RT-PCR (Fig. 7B). The results showed that changes in lncRNAs expression levels were consistent with RNA-seq data. Moreover, we also tested the reported lncRNAs, and the results were consistent with the literature (Fig. 7B, 7C).

Validation of differentially expressed lncRNAs and reported lncRNAs by RT-qPCR and RT-PCR. p < 0.05 (*), < 0.01 (**), and < 0.001 (***) were considered statistically significant. A Validation of differentially expressed lncRNAs by RT-qPCR, B validation of differentially expressed lncRNAs and reported lncRNAs by RT-PCR and C validation of reported lncRNAs by RT-qPCR.
Discussion
Traditional studies of host cell responses to influenza infection have mainly focused on protein-coding genes. Recently, several studies have shown that lncRNAs regulate virus replication by mediating host gene expression. For example, the lncRNA NRAV negatively regulates the initial transcription of multiple critical interferon-stimulated genes by affecting histone modification and significantly promotes IAV replication (Ouyang et al.2014). Additionally, the lncRNA NEAT1, which is induced by influenza virus, is essential for the formation of nuclear body paraspeckles and thereby facilitates the expression of antiviral genes, including cytokines such as interleukin-8 (IL-8), probably through relocating transcriptional regulator splicing factor proline/glutamine-rich (a NEAT1-binding paraspeckle protein) from the IL-8 promoter to the paraspeckles (Imamura et al.2014). Previous studies have also shown that the lncRNA Bst2/BISPR, which is induced by an influenza virus lacking NS1, is unable to block the interferon response. This lncRNA is located in genome close to BST and positively regulates the expression of BST2, but does not affect the expression of other genes adjacent to BST2 (Barriocanal et al.2014). VIN, a large intergenic lncRNA induced by several IAV strains, including H1N1, H3N2, and H7N7, promotes influenza virus replication (Winterling et al.2014). In this study, we found that 6129 lncRNAs and 50,031 mRNA transcripts were differentially expressed. Because several lncRNAs have been identified as modulators in virus-induced host responses, we predicted that these lncRNAs may play crucial roles in regulating the host response to IAV, although the mechanisms require further exploration.
Notably, in this study, the functions of lncRNAs were predicted according to their cis- or trans-target genes. GO terms were significantly enriched in biological processes, such as RNA processing, cell cycle, and cellular metabolic process, suggesting that lncRNAs induced by H3N2 infection may regulate metabolism and affect H3N2 replication. To date, several studies have suggested that viruses regulate host metabolism to affect virus replication. Influenza virus manipulates sphingolipid metabolism and increases the level of sphingosine kinase 1 to promote virus replication (Vijayan and Hahm 2014). Human cytomegalovirus (HCMV) requires glycolysis for replication of the viral genome (McArdle et al.2011), and rhinovirus infection results in extensive alterations in cellular metabolism essential for viral replication, including enhancing utilization and uptake of glucose as well as enhancing nucleotide synthesis and lipogenesis (Gualdoni et al.2018). HCMV and herpes simplex virus-1 (HSV-1) both induce major metabolic alterations in their host cells, and HCMV profoundly enhances TCA compound levels and alters pathways that generate substrates for lipid metabolism essential for its replication. Moreover, HSV-1 upregulates pyrimidine nucleotide biosynthesis for its replication (Vastag et al.2011). LncRNAs regulate metabolic enzymes to affect virus replication. The lncRNA ACOD1 is induced in cells infected with various viruses and can bind the metabolic enzyme glutamic-oxaloacetic transaminase, increased its catalytic activity, and facilitate the production of metabolites that promote viral propagation (Wang et al.2017).
In addition to metabolism, KEGG pathway analysis showed that target genes of differentially expressed lncRNAs were enriched in autophagy. IAV infection plays complex roles in regulating autophagy. Indeed, IAV induces the formation of autophagosomes in rapamycin-treated Madin-Darby canine kidney cells (Tanida et al.2008; Zhou et al.2009). Additionally, several viral proteins are involved in the induction of autophagy. For example, M2 protein alone is able to induce the initial steps of autophagosome formation (Gannage et al.2009; Zhou et al.2009; Zhirnov and Klenk 2013), viral HA protein can slightly activate autophagy, and NS1 induces autophagy indirectly by promoting the synthesis of HA and M2 proteins (Zhirnov and Klenk 2013). Moreover, IAV infection inhibits the degradation of autophagosomes by blocking their fusion with lysosomes (Gannage et al.2009). Therefore, IAV likely regulates autophagy through lncRNAs.
In summary, in this study, we analyzed the expression profiles of lncRNAs in A549 cells infected by H3N2 for the first time. In total, 6129 lncRNAs were differential expressed in H3N2-infected A549 cells compared with that in normal cells. These results indicated that lncRNAs played regulatory roles in metabolism, autophagy, and other related pathways following H3N2 infection. Further studies will be needed to determine the roles of these lncRNAs play in H3N2 infection, and that may lead to novel vaccines developments for preventing IAV.
References
Barriocanal M, Carnero E, Segura V, Fortes P (2014) Long non-coding RNA BST2/BISPR is induced by IFN and regulates the expression of the antiviral factor tetherin. Front Immunol 5:655
Carpenter S (2016) Long noncoding RNA: novel links between gene expression and innate immunity. Virus Res 212:137–145
Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, O’Neill LA, Moore MJ, Caffrey DR, Fitzgerald KA (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science 341:789–792
Costa FF (2010) Non-coding RNAs: meet thy masters. BioEssays 32:599–608
Dangi T, Jain A (2012) Influenza virus: a brief overview. Proc Natl Acad Sci India Sect B Biol Sci 82:111–121
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Feng M, Yuan Z, Xia W, Huang X, Wang X, Yan Y, Liao M, Zhou J (2018) Monoclonal antibody against the universal M2 epitope of influenza A virus. Appl Microbiol Biotechnol 102:5645–5656
Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia-Sastre A, Munz C (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380
Gualdoni GA, Mayer KA, Kapsch AM, Kreuzberg K, Puck A, Kienzl P, Oberndorfer F, Fruhwirth K, Winkler S, Blaas D, Zlabinger GJ, Stockl J (2018) Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc Natl Acad Sci USA 115:E7158–E7165
Horimoto T, Kawaoka Y (2005) Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 3:591–600
Ilott NE, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, Goodhead I, Hertz-Fowler C, Heger A, Hall N, Donnelly LE, Sims D, Lindsay MA (2014) Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun 5:3979
Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y, Sekimizu K, Akimitsu N (2014) Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 53:393–406
Ito T, Couceiro JN, Kelm S, Baum LG, Krauss S, Castrucci MR, Donatelli I, Kida H, Paulson JC, Webster RG, Kawaoka Y (1998) Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J Virol 72:7367–7373
Jiang M, Zhang S, Yang Z, Lin H, Zhu J, Liu L, Wang W, Liu S, Liu W, Ma Y, Zhang L, Cao X (2018) Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 173(906–919):e913
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345–W349
Lemon SM, Mahmoud AA (2005) The threat of pandemic influenza: are we ready? Biosecur Bioterror 3:70–73
Li A, Zhang J, Zhou Z (2014) PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform 15:311
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
McArdle J, Schafer XL, Munger J (2011) Inhibition of calmodulin-dependent kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J Virol 85:705–714
Nishitsuji H, Ujino S, Yoshio S, Sugiyama M, Mizokami M, Kanto T, Shimotohno K (2016) Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc Natl Acad Sci USA 113:10388–10393
Ouyang J, Zhu X, Chen Y, Wei H, Chen Q, Chi X, Qi B, Zhang L, Zhao Y, Gao GF, Wang G, Chen JL (2014) NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 16:616–626
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295
Qiu L, Wang T, Tang Q, Li G, Wu P, Chen K (2018) Long non-coding RNAs: regulators of viral infection and the interferon antiviral response. Front Microbiol 9:1621
Simonsen L, Clarke MJ, Williamson GD, Stroup DF, Arden NH, Schonberger LB (1997) The impact of influenza epidemics on mortality: introducing a severity index. Am J Public Health 87:1944–1950
Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K (1998) Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J Infect Dis 178:53–60
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166
Szczesniak MW, Makalowska I (2016) lncRNA-RNA interactions across the human transcriptome. PLoS ONE 11:e0150353
Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. Methods Mol Biol 445:77–88
Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K (2003) Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186
Tong L, Qiu Y, Wang H, Qu Y, Zhao Y, Lin L, Wang Y, Xu W, Zhao W, He H, Zhao G, Zhang MH, Yang D, Ge X, Zhong Z (2019) Expression profile and function analysis of long non-coding RNAs in the infection of coxsackievirus B3. Virol Sin. https://doi.org/10.1007/s12250-019-00152-x
Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD (2011) Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog 7:e1002124
Vijayan M, Hahm B (2014) Influenza viral manipulation of sphingolipid metabolism and signaling to modulate host defense system. Scientifica (Cairo) 2014:793815
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W (2013) CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res 41:e74
Wang P, Xu J, Wang Y, Cao X (2017) An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 358:1051–1055
Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y (1992) Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179
Winterling C, Koch M, Koeppel M, Garcia-Alcalde F, Karlas A, Meyer TF (2014) Evidence for a crucial role of a host non-coding RNA in influenza A virus replication. RNA Biol 11:66–75
Wu X, Wang H, Bai L, Yu Y, Sun Z, Yan Y, Zhou J (2013) Mitochondrial proteomic analysis of human host cells infected with H3N2 swine influenza virus. J Proteomics 91:136–150
Xue J, Chambers BS, Hensley SE, Lopez CB (2016) Propagation and characterization of influenza virus stocks that lack high levels of defective viral genomes and hemagglutinin mutations. Front Microbiol 7:326
Zhirnov OP, Klenk HD (2013) Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J Virol 87:13107–13114
Zhou Z, Jiang XJ, Liu D, Fan Z, Hu XD, Yan JG, Wang M, Gao GF (2009) Autophagy is involved in influenza A virus replication. Autophagy 5:321–328
Acknowledgements
This study is supported by grants from the National Key technology R&D Program of China (Grant No. 2015BAD12B01), the China Agriculture Research System (Grant No. CARS-40-K13) and the National Science Foundation of China (Grant No. 31502084).
Author information
Authors and Affiliations
Contributions
JZ and YZ designed the experiments and wrote the paper. YZ performed the majority of the experiments. TY and YD analyzed the data. YL prepared the sequencing samples. JL verified sequencing results and BH edited pictures.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This article does not contain any studies with human or animal subjects.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Yu, T., Ding, Y. et al. Analysis of Expression Profiles of Long Noncoding RNAs and mRNAs in A549 Cells Infected with H3N2 Swine Influenza Virus by RNA Sequencing. Virol. Sin. 35, 171–180 (2020). https://doi.org/10.1007/s12250-019-00170-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-019-00170-9