
Abstract
Over the past few years, cellular immunotherapy has emerged as a novel treatment option for certain forms of hematologic malignancies with multiple CAR-T therapies now routinely administered in the clinic. The limitations of generating an autologous cell product and the challenges of toxicity with CAR-T cells underscore the need to develop novel cell therapy products that are universal, safe, and potent. Natural killer (NK) cells are part of the innate immune system with unique advantages, including the potential for off-the-shelf therapy. A recent first-in-human trial of CD19-CAR-NK infusion in patients with relapsed/refractory lymphoid malignancies proved safe with promising clinical activity. Building on these encouraging clinical responses, research is now actively exploring ways to further enhance CAR-NK cell potency by prolonging in vivo persistence and overcoming mechanisms of functional exhaustion. Besides these strategies to modulate CAR-NK cell intrinsic properties, there are increasing efforts to translate the successes seen in hematologic malignancies to the solid tumor space. This review will provide an overview on current trends and evolving concepts to genetically engineer the next generation of CAR-NK therapies. Emphasis will be placed on innovative multiplexed engineering approaches including CRISPR/Cas9 to overcome CAR-NK functional exhaustion and reprogram immune cell metabolism for enhanced potency.
Similar content being viewed by others

Introduction
Adoptive cellular immunotherapy has moved from the bench to the bedside and heralds a new, promising era in the treatment of hematologic malignancies, with multiple CD19-directed chimeric antigen receptor (CAR)-T therapies now available [1,2,3,4,5,6]. With the recent FDA approval of CAR-T cells targeting B cell maturation antigen (BCMA) in patients with relapsed/refractory (R/R) multiple myeloma [7, 8], the therapeutic scope of adoptive cellular therapeutics has been successfully expanded beyond CD19 positive lymphoid malignancies. While CAR-T cell products undoubtfully have improved clinical outcomes and provided new hope for patients with refractory and relapsed hematologic malignancies, nearly 5 years of real-world clinical experience since market approval of the first CAR T cell product have also led to a somewhat sobering experience for many in the field. Besides the cumbersome logistics associated with autologous cell manufacturing and high rates of cell therapy-related toxicities, the most pressing concern remains suboptimal long-term disease control. As the most recent results from the initial pivotal trials show, current generation CAR-T products still fail to deliver long-term responses for many patients, with a significant proportion of treated patients eventually relapsing and succumbing to their disease [9,10,11].
CAR constructs are generally composed of an extracellular synthetic receptor for antigen recognition, an extracellular hinge region, a transmembrane domain and one or multiple intracellular co-stimulatory domains. The CAR receptor typically is derived from a murine mAb in the form of a single-chain variable fragment (scFv) but can also be encoded to express a naturally occurring surface receptor with affinity for tumor-specific ligands. The latter approach has gained increased attention as it circumvents the alloreactive potential arising from the murine scFv origin which can lead to CAR immunogenicity and result in treatment failure [12]. Depending on the composition of the intracellular signaling domains, CAR constructs can be ascribed to certain generations. While first generation CAR constructs rely on a single intracellular signaling domain [13], second [14, 15] and third [16, 17] generation CAR molecules are designed for dramatically augmented intracellular effector signaling by carrying either one or multiple additional co-stimulatory endodomains, typically in the form of either CD28 or 4-1BB.
In recent years, CAR engineering of natural killer (NK) cells has gained considerable traction due to the unique biological properties of NK cells that make them highly attractive cells for cancer immunotherapy. NK cells are innate immune cells which mediate cytotoxicity against cancerous and virally infected cells through multiple mechanisms of action [18]. The traditional dogma, by which NK recognition of abnormal cells is primarily driven by loss of target MHC-I surface expression, has over time been expanded by a more subtle understanding of the complex integration of multiple stimuli from both activating and inhibitory receptors which collectively determine NK cell activation state and target engagement. NK cells, even when genetically engineered to express a CAR molecule, retain their intrinsic capacity to recognize tumor cells through their native receptors, thereby providing them with an additional layer of anti-tumor reactivity independent of CAR-redirected killing. Of note, NK cells do not rely on the T-cell receptor (TCR) for cytotoxic killing. This endows them with a more favorable safety profile compared to their T-cell contenders, which, in the allogeneic setting, need to be further genetically modified to circumvent graft versus host disease (GvHD).
CAR-NK cells can be generated from NK cells derived from a variety of different sources including cord blood (CB), peripheral blood cell (PB), NK cell lines as well as hematopoietic and induced pluripotent stem cells (iPSC). While each source of NK cell presents its unique advantages and challenges, these have been reviewed in detail previously [19, 20] and will not be the focus of this article.
A new horizon—translating CAR-NK immunotherapy into the clinic
Clinical application of CAR-engineered NK cells was first reported in 2018 by a group from mainland China. Using a third-generation CD33-directed CAR construct with CD28 and 4-1BB co-stimulatory domains to transduce NK-92-derived NK cells, the researchers were able to demonstrate in a phase 1 study (NCT02944162) the safe infusion of up to 5 × 109 CAR-NK cells into three patients with R/R acute myeloid leukemia (AML). While the therapy proved safe and without any evidence of significant adverse events, no durable remissions were attained. The study’s negative results were attributed mainly to the limited in vivo persistence of irradiated NK-92 cells, with CAR-NK levels dropping below detection thresholds within 1 week after infusion [21]. Indeed, in vivo expansion and long-term persistence of CAR-engineered lymphocytes constitute the linchpin of cell-based therapies which, when achieved, provide a decisive edge over mAb therapies by contributing to durable clinical responses. NK cells physiologically have a life span of only 2 weeks [22] which requires any successful CAR-NK engineering approach to overcome this inherent biological boundary, to achieve sustained responses.
Our group successfully pioneered a novel cord-blood-derived CD19-directed CAR-NK product with constitutive IL-15 cytokine support (CD19-CD28-ζ-2A-iCasp9-IL15) for sustained in vivo persistence [23]. In an interim analysis of the first 11 patients with heavily pre-treated R/R lymphoid malignancies who were infused with this product, eight (73%) achieved clinically meaningful responses with seven (63%) achieving a complete remission (CR) (NCT03056339) [24]. The interim results demonstrated an excellent safety profile with no occurrences of cytokine release syndrome (CRS), neurological toxicities, or GvHD. Of note, CAR-NK cells were detected for up to 1 year after infusion with responders exhibiting significantly enhanced CAR-NK peak copy numbers compared to patients who did not have a response. While larger multicenter trials will eventually need to validate our encouraging findings and assess long-term response duration, the interim results clearly demonstrate the potential of CAR-NK therapy for patients with high-risk lymphoid malignancies.
Most recently, preliminary data from an ongoing phase 1 clinical trial (NCT04023071) investigating an induced pluripotent stem cell (iPSC)-derived NK cell product, FT516, have been reported. FT516 has been engineered to express a non-cleavable Fc receptor (CD16a) to specifically redirect NK cells using monoclonal antibodies and enhance antibody-dependent cellular cytotoxicity (ADCC) [25]. Among the 11 patients with R/R B-NHL who received the investigational cell therapy in combination with rituximab, eight patients achieved an objective response (73%), six patients (55%) attained a complete remission and no evidence of CRS, ICANS or GvHD was observed [26]. Longer follow-up data will need to address whether the observed responses are durable and infused NK cells persist over time.
Evolving clinical trials landscape for CAR-NK immunotherapy
The past few years have witnessed an unprecedented acceleration in the early phase clinical trial activity in CAR-NK immunotherapy with multiple CAR-modified NK cell products currently under clinical investigation. Across the trials landscape, investigators are testing CAR constructs against a multitude of target antigens, using different sources of NK cells and various modular CAR designs. Overall, there are two main trends to highlight. Most noticeably, there has been a shift in the scope of targeted disease. While most of the earlier trials focused on targeting hematologic malignancies, many of the more recent trials seek out to explore CAR-NK cell immunotherapy as a new means to combat solid tumors. Second, there is an increasing appreciation of more sophisticated construct designs, enabled by multiplexed gene editing approaches, to enhance NK cell potency and persistence. In this review we first will summarize the recent clinical trial activity, highlighting the most promising CAR-NK cell candidates currently in clinical testing. In the second part of this review, we will elaborate on some of the novel genetic engineering avenues pursued to devise the next generation of CAR-NK cell immunotherapies.
Table 1 provides a list of all currently registered CAR-NK cell trials. While most CAR-based therapeutic approaches deliberately set out to target an individual tumor surface antigen in a highly specific manner, a different strategy has been employed with the design of NKX101, a donor-derived CAR-modified NK cell product engineered to express a synthetic NKG2D activating receptor [27]. NKG2D, a germline-encoded NK cell receptor, interacts with a plethora of stress ligands expressed on abnormal cells and is considered one of the central activating receptors driving innate NK cell effector responses. NKX101 (NGD2D-OX40-CD3ζ-Il15) demonstrated promising anti-leukemic activity in vivo [27] and is currently being evaluated in a phase 1 trial in patients with R/R AML as well as those with high-risk myelodysplastic syndrome (NCT04623944).
Another CAR-NK candidate which expresses both non-cleavable CD16a and a CD19-directed CAR receptor (CAR.19-NKG2D-2B4-CD3ζ-IL15RF-hnCD16) is currently evaluated clinically in advanced B-NHL and CLL in combination with anti-CD20 agents (NCT04245722). While no interim reports have yet been reported, preclinical in vivo data suggest superior tumor cell clearance for the multiplexed CAR candidate versus primary CD19-CAR-T cells [28]. FT538, a further CAR-NK candidate in clinical testing, has been engineered to be resistant to anti-CD38-mediated fratricidal killing by means of double allelic knock-out of CD38 and is being investigated against R/R multiple myeloma in combination with Daratumumab (NCT04614636). In contrast to CD38 wild type therapeutics, CD38−/− CAR-NK cells may provide a clinical edge as they could eventually move into earlier lines of myeloma therapy including daratumumab-naïve patients.
Other ongoing studies are evaluating CB-derived NK cells CD19 (NCT04796675) and SARS-CoV-2 (NCT04324996), and NK-92-derived CAR-NK cells across a broad spectrum of tumor antigens including CD7 (NCT02742727), CD19 (NCT02892695), BCMA (NCT03940833), HER2 (NCT03383978) and PD-L1 (NCT04847466). While none of these trials have reported interim results yet, it will be interesting to observe the efficacy of CAR-NK cells against non-lymphoid malignancies.
Building the next generation of CAR-NK therapies
While the first part of this review has focused on the ongoing clinical development of CAR-NK immunotherapy, the second part aims to shed light on the most recent preclinical efforts to overcome some of the shortcomings seen with the current generation cell therapies.
Cancer cells have evolved sophisticated immunosuppressive mechanisms to thwart the effects of infiltrating lymphocytes and evade tumor immune surveillance. While earlier research focused predominantly on cancer-intrinsic mechanisms of immune escape, there has been an increasing appreciation of immune cell specific factors which underpin the fundamental phenotypic reconfigurations which leave immune cells in a dysfunctional state of ineffective tumor control. Finding a way to make CAR-engineered NK cells more potent, without compromising their favorable safety profile, is critical for the next generation of cell therapies. Current CAR-NK therapy constraints revolve around three recurring themes. First, CAR-NK therapies have thus far only been successfully applied to a very narrow repertoire of cancer-specific antigens. Second, limited in vivo persistence and eventual immune cell exhaustion pose a significant hurdle for long-term efficacy. Lastly, impaired trafficking to tumor beds and the delicacies of maneuvering the hostile tumor microenvironment impede the effective interaction of NK cells and their adversaries, ultimately leading to tumor immune evasion.
In the following sections of this review, we will (i) provide an overview of the ongoing preclinical efforts to redirect CAR specificity to extend the therapeutic scope of CAR-NK therapies and (ii) discuss some of the most interesting concepts to genetically reprogram CAR-NK cells to overcome some of the longstanding hurdles outlined above. We will then highlight the emerging applications of CAR-NK immunotherapy beyond hematologic malignancies including against COVID-19 and solid tumors, and iii) end this synopsis with an outlook on how we perceive CAR-NK immunotherapy will impact the field of cellular therapy and hematology/oncology over the coming years.
Extending the therapeutic scope for CAR-NK immunotherapy in hematologic malignancies
Most of the initial work in CAR-NK cell-based immunotherapy has focused on a very limited set of target antigens, mainly in the liquid cancer sphere. This next section will focus on the quest to find suitable tumor antigens to address cancer entities beyond B lymphoid malignancies and broaden the therapeutic scope for CAR-NK cell immunotherapy.
Aggressive T-cell malignancies
Aggressive T-cell malignancies represent an area of high unmet medical need, with dismal clinical outcomes and very limited therapeutic options at hand. CAR-T-based approaches have proven inherently challenging as shared expression of surface antigens among CAR-engineered T cells and transformed T lymphoblasts poses the risk of fratricide, a phenomenon by which infused CAR-T cells start attacking themselves leading to their own elimination. Another reason for concern is rooted in the potential contamination of autologous T-cell products with malignant T cells during the manufacturing process. CAR-NK cells lack expression of T-cell markers and can be derived from allogeneic sources, providing a compelling edge in the treatment of T-cell malignancies.
One group tested NK-92-derived cells, transduced to express a third-generation CAR construct (CD28-4-1BB-CD3ζ) against the pan-T-cell antigen CD3 and achieved significantly reduced tumor burden and prolonged survival in a T-ALL mouse model [29]. Although not as broadly expressed as CD3, CD4 represents another potential antigen for T-cell-directed therapies. NK-92 cells transduced with a third-generation anti-CD4-CAR construct (4-1BB-CD28-CD3ζ), when tested against a xenograft mouse model of anaplastic large cell lymphoma (ALCL), resulted in significant tumor control and prolongation of survival [30]. CD5 has similarly been targeted with another group engineering NK-92 cells with a third-generation CD5-CAR construct (4-1BB-CD28-CD3ζ) which resulted in enhanced anti-tumor efficacy in a T-ALL mouse model. More recently, a group from Tianjin, China designed a second-generation CD5-directed CAR construct (2B4-CD3ζ) which showed specific cytotoxicity against primary T-cell lymphoma cells in vitro and improved survival in a T-ALL in vivo mouse model [31]. CD7, while non-essential for normal T-cell development and homeostasis, is found on the majority of T-ALL clones and provides another alluring target for CAR receptor design. You et al. were able to construct a third-generation CAR molecule (4-1BB-CD28-CD3ζ) based on a CD7 nanobody which elicited potent anti-lymphoma responses in a mouse model of aggressive T-cell leukemia without significant fratricide [32]. Clinically, CD7-CAR-NK cells are already under investigation against aggressive T-cell malignancies in a Chinese-led phase 1 trial (NCT02742727).
While fratricide and contamination are of lesser concern with NK cell-based products, prolonged T-cell aplasia due to inadvertent elimination of healthy T cells resulting in severe immunosuppression, continues to be a major obstacle to be resolved before T-cell-directed therapies become a viable option in the clinic [33].
One strategy to mitigate the adverse effects of long-term T-cell aplasia inherent to T-cell-directed CAR-modified immune cells is to restore the cytotoxic potential of non-engineered NK cells using bispecific engagers that specifically guide NK cells towards tumor cells and mediate NK activation through high-affinity binding of their CD16A receptor. Our group has reported on the first successful application of pre-complexed NK cells using the tetravalent bispecific CD16A:CD30 immune cell engager AMF13 to induce CAR-like responses against CD30 positive malignancies. In brief, cord-blood-derived NK cells were pre-conditioned with the cytokines IL-12, IL-15 and IL-18 and pre-complexed with AMF13. Pursuing this approach, we were able to demonstrate increases in the cytotoxic potential of AFM13-loaded NK cells in vitro. When tested in lymphoma-bearing mice, AFM13-complexed NK cells elicited markedly improved anti-tumor responses and significantly prolonged survival, while no significant adverse toxicities were observed [34]. This approach is currently being tested in the clinic (NCT04101331).
Acute myeloid leukemia
Engineering CAR constructs targeting AML has proven challenging with two obstacles standing out [35]. Shared antigen expression of leukemic blasts and hematopoietic progenitor cells, as is the case for CD123 and CD33, raises the potential threat of on-target off-tumor toxicities eliciting prolonged multi-lineage cytopenias. Clonal heterogeneity and varied antigen expression across different AML populations represent another challenge leaving single-antigen targeted approaches prone to disease relapse, particularly in the presence of distinct leukemia-initiating clones characterized by discordant antigen signatures compared to bulk leukemic cells [35]. Our group and others have devised CAR molecules targeting CD123 using both third-generation (CD28-4-1BB-CD3ζ) and fourth-generation (CD28-CD3ζ-IL15) construct designs, and provided encouraging evidence for anti-leukemia cytotoxicity in vitro including against primary AML blasts [36, 37]. More recently, a third-generation CAR construct targeting CD4 (CD28-4-1BB-CD3ζ) was shown to specifically eliminate primary CD4 positive AML blasts and suppress disease progression in a cell-line-derived xenograft mouse model [38]. An innovative approach using the novel site-directed gene-insertion CRISPaint technology (CRISPR-assisted insertion tagging) to engineer two third-generation CD33-directed CAR-NK candidates (CD4-4-1BB-CD3ζ and NKG2D-2B4-CD3ζ) has been reported to elicit anti-leukemic activity in vitro [39].
An ingenious strategy to generate logic-gated CAR-NK circuits to address the two longstanding challenges, heterogeneous and shared antigen expression, which have thus far held back the success of AML cellular therapeutics, was reported by Dr. Gary Lee’s group [40]. Guided by a data-driven bioinformatics approach to predict optimal antigen combinations, they devised logic-gated CD33-OR-FLT3-NOT-EMCN CAR-NK cells. These cells specifically target AML blasts and leukemia-initiating cells by recognizing both CD33 and FLT3, but are inhibited when encountering endomucin (EMCN) which is present on up to 70% of FLT3-expressing hematopoietic stem cells. When co-cultured with FLT3 + EMCN-AML blasts and healthy FLT3 + EMCN + hematopoietic progenitor cells, their investigational allogeneic CAR-NK candidate exhibited selective and differential killing of the targeted blasts while sparing the hematopoietic stem cell compartment [41, 42].
Multiple myeloma
Multiple myeloma cells express a number of NK activating ligands and hence should be uniquely positioned to be targeted harnessing the power of CAR-modified NK cells [43]. Reports of CAR-NK immunotherapy to combat multiple myeloma date back to 2014 with one group reporting on the in vitro efficacy of a CD138-directed CAR construct used to genetically modify NK-92MI cells. Another group endowed NK-92 cells with a CS1-targeted CAR receptor which prolonged survival in vivo in a xenograft mouse model [44, 45]. Early efforts to target plasma cell diseases using NK cells were, however, restrained by concerns over NK fratricide due to their shared expression of CS1 and CD38. Independent of this NK cell intrinsic constraint, the field has shifted its attention towards exploring novel antigens, including BCMA, with the potential to induce remissions in patients with previous exposure to anti-CD38 agents. Reinforcing this notion, a two second-generation NK-92MI-CAR candidates (4-1BB-CD3ζ) targeting either NKG2D or BCMA were reported to mediate potent anti-myeloma activity in vitro [46,47,48]. Another team similarly was able to observe in vitro anti-tumor efficacy using a backbone encoding for both non-cleavable CD16a and BCMA combined with double allelic CD38 knock-out, paving the way for future combination therapies with CD38-targeted agents [49].
The next frontier—multiplexed genetic editing
Rational construct design
Besides redirecting CAR-NK cells to different target antigens, more recently the research focus has increasingly shifted towards optimized construct design based on modular combinations of different transmembrane and intracellular costimulatory domains to enhance CAR-NK potency.
Empowered by the versatile genetic engineering capabilities available today, researchers are now set up to fine-tune CAR constructs to induce more potent anti-tumor responses, increase antigen affinity or prolong in vivo persistence. Multiple co-stimulatory elements have been investigated for their potential to mount more effective anti-tumor responses, including domains derived from the immunoglobulin superfamily (CD28, ICOS), the TNF receptor superfamily (4-1BB, CD27, OX40, and CD40) and others including CD40L and toll-like receptors (TLRs) [50]. Compared to some of the earlier CAR-NK constructs which were primarily based on co-stimulatory domains involved in T-cell activation, there has been an increasing appreciation for NK-specific signaling adaptors with DAP10, DAP12 and 2B4 taking on particularly prominent roles [51,52,53]. These intracellular signaling adaptors mediate NK cell activation upon association with their respective upstream receptors. In a preclinical study investigating a CD19-directed CAR-NK candidate, incorporation of DAP10, the physiological adaptor for NK activating receptor NKG2D, enhanced the anti-tumor potency over a construct using CD3ζ signaling alone. Similar observations have been reported by others with amplified anti-tumor potency by including DAP12 in a prostate stem cell antigen (PSCA)-targeted CAR construct [54] and another group employing a 2B4-enhanced signaling strategy in mesothelin-targeted iPSC-derived natural killer cells [55].
Armored CARs
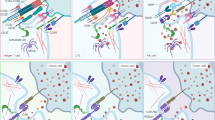
Modifications to CAR construct design across the first three CAR generations rely on physiologically occurring immune cell receptor domains. Fourth-generation constructs, dubbed armored CARs, take a dramatically different approach by incorporating molecular payloads and thereby endowing the CAR-modified immune cells with additional features and functionality which cannot be found within any physiological immune cell receptor. This approach enables CAR constructs to be engineered to address some of the inherent biological limitations of cellular immunotherapy, thereby positioning CAR engineering in the center of the synthetic biology revolution (Fig. 1).

Evolution of CAR construct design. (1) First generation CAR molecules contain a synthetic extracellular receptor for target antigen recognition, a transmembrane domain and one intracellular signaling domain. (2,3) Second and third generation CAR constructs are endowed with one or more intracellular co-stimulatory domains for augmented signaling. (4) Fourth generation constructs, dubbed armored CARs, contain molecular payloads which endow CAR-engineered cells with additional features and functionality. (5) Multi-specific targeting refers to genetic engineering strategies which aim to simultaneously target multiple tumor antigens. (6) Logic-gated CAR molecules are engineered to express both an activating (aCAR) and inhibitory (iCAR) CAR receptor on their surface. The former recognizes tumor-specific epitopes and is able to initiate immune cell activation and target engagement. The latter detects epitopes exclusively present on healthy tissues and inhibits immune cell activation. By integrating both signals, logic-gated CAR-modified NK cells can differentially kill tumor cells while sparing healthy tissues with shared antigens. (7) Bispecific Killer engagers (BiKes) are bispecific antibodies with binding sites for both FcRγIII (CD16A) and a tumor-specific epitope. Acting as a molecular glue, BiKes can bind and bring NK cells in close proximity of tumor cells for specific target elimination in a CAR-like manner. aCAR, activating chimeric antigen receptor; GzmB, granzyme B; HSC, hematopoietic stem cell; iCAR, inhibitory chimeric antigen receptor; IFNγ, interferon gamma; Il-15, interleukin 15; mAb, monoclonal antibody; PFN, perforin; T-NHL, T non-Hodgkin lymphoma; TNFα, tumor-necrosis factor alpha. Image created in BioRender
Arguably the most pressing biological constraint is limited in vivo persistence in the absence of exogenous cytokine support. While the clinical administration of cytokines is feasible, it carries with it considerable safety concerns and might inadvertently stimulate immunosuppressive immune cell compartments [56]. To overcome these obstacles, our group demonstrated how constitutive transgenic IL-15 cytokine support enables sustained in vivo persistence and augmented anti-tumor efficacy in a CD19-targeted CAR-NK candidate in patients with lymphoid malignancies [23, 24]. Empowered by modern bi- and multicistronic vector platforms, fourth-generation constructs can contain more than one desirable genetic modification, an engineering approach referred to as multiplexed genetic engineering. In the case of our group’s CD19-redirected CAR-NK product, we additionally included a molecular safety switch using an inducible Caspase9 (iCasp9) which can be activated by administration of a small molecule dimerizing drug to eliminate all circulating adoptive NK cells, in the event of adverse toxicities [23, 24].
Genetic disruption of CAR-NK immune checkpoints
Despite the remarkable successes seen with the administration of adoptive NK cell-based therapy, immune cell exhaustion remains a therapeutic barrier and strategies to overcome this are warranted to enhance long-term in vivo anti-tumor efficacy. For building the next generation of CAR-NK therapies, basic research on fundamental NK cell biology has once again shifted to the center of attention and significant efforts are underway to identify negative regulators of NK cell immune function. Recent advances in the understanding of the transcriptional networks involved in immune cell exhaustion, in large extent driven by high-throughput single-cell-based studies, have helped establish distinct signatures of dysfunctional NK cells. Several distinct phenotypic changes have been proposed to delineate exhausted NK cell subsets with upregulation of PD-1, LAG-3, TIM-3 [57] as well as TIGIT and KLRG1 [58] among those markers that confer impaired cytotoxic potential and hence embody putative candidates for targeted genetic ablation.
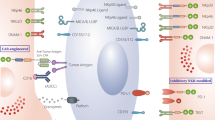
While the emergence of highly customizable and powerful viral vector platforms to deliver CAR molecules into various immune cell subsets was a prerequisite to enable the extraordinary strides seen in cellular immunotherapy over the last decade, the increasing adoption of targeted genetic perturbations using CRISPR/Cas9-mediated gene editing has opened up entirely new routes for modulating the underlying immune cell biology. Multiplexed genetic engineering strategies in particular allow for further improvement of CAR-modified immune cells in a site-directed manner so as to disrupt molecular pathways that negatively impact immune cell function. Multiple groups, including our own, have embarked on this journey to engineer the next generation of CAR-NK immunotherapies with enhanced potency, increased in vivo persistence and resistance to mechanisms of functional exhaustion with the ultimate goal to upend the field of cell therapy as we know it today (Fig. 2).

Multiplexed engineering strategies for building the next-generation of CAR-NK immunotherapy. (1) Ectopic expression of chemokine receptors enhances NK trafficking and infiltration into tumor beds. (2) Genetic disruption of negative regulators of NK immune function can help overcome functional exhaustion. (3) Neutralizing scFvs can intercept extracellular GM-CSF which has been linked to CRS/ICANS. (4) Cytokine pre-conditioning using Il-12/15/18 induces formation of memory-like NK cells with enhanced potency. (5) Knock-out of both surface and intracellular molecules conditions NK cells for combination therapies by preventing fratricide (CD38 KO) or immunosuppression (GR KO). (6) Metabolic reprogramming aims to render NK cells resistant to mechanisms of functional exhaustion by intervening in metabolic pathways. (7) Targeted genetic ablation of inhibitory receptors can safeguard CAR-NK cells against the immunosuppressive tumor microenvironment. A2AR, adenosine A2A receptor; CRS, cytokine release syndrome; GM-CSF, granulocyte macrophage colony stimulating factor; GR, glucocorticoid receptor; ICANS, immune cell-associated neurotoxicity syndrome; Il-10/12/15/18, interleukin 10/12/15/18; IDO, indoleamine 2,3-dioxygenase; PGE2, prostaglandin E2; scFv, single-chain variable fragment; TGFβ, transforming-factor beta; TGFBR2, transforming-factor beta receptor 2. Image created in BioRender
Genetic targeting of inhibitory immune checkpoints has emerged as a tantalizing concept to disrupt negative regulators of CAR-NK function for enhanced anti-tumor control. While NK cell activation states are intricately controlled by the complex interplay of both activating and inhibitory surface receptors, checkpoint blockade aims to skew this equilibrium towards heightened anti-tumor immunity. Genetic deletion of NKG2A, one of the most prominent germline-encoded inhibitory NK cell receptors, has previously been linked to increased cytotoxic potential against tumors expressing its cognate ligand HLA-E [59]. Blockade of TIGIT, another inhibitory checkpoint essential for maintaining balanced immune responses, has similarly been shown to prevent NK cell exhaustion [60]. Our group successfully targeted the cytokine-inducible Src homology 2-containing (CIS) protein, a key cytokine checkpoint upstream of interleukin 15 signaling, and achieved enhanced metabolic fitness and effector function in armored cord-blood-derived CAR-NK cells [61]. Other groups are striving to translate the lessons learned in the context of T-cell immunobiology and are investigating the effects of PD-1/PD-L1 and CTLA-4 blockade on NK cells [62, 63].
Safeguarding against the immunosuppressive tumor microenvironment
Apart from NK-intrinsic immune checkpoints, there are several extrinsic factors arising from the hostile tumor microenvironment which collectively create a highly unfavorable milieu for infiltrating immune cells. A mix of nutrient deprivation, acidity and hypoxia deprives infiltrating effector cells their anti-tumor activity. Furthermore, tumor cells have evolved mechanisms to attract regulatory immune cell compartments such as regulatory B cells and regulatory T cells as well as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), platelets, fibroblasts and stromal cells which secrete immunosuppressive cytokines including transforming growth factor β1 (TGFβ), adenosine, indoleamine 2,3-dioxygenase (IDO) and prostaglandin E2 (PGE2), further obliterating any remnants of an effective anti-tumor response. One of these extracellular immunosuppressive metabolites, adenosine, has successfully been rendered ineffective by blocking the expression of A2A adenosine receptor (A2AR) on the surface of NK cells. A2AR-deficient NK cells exhibited enhanced tumor control in a xenograft mouse model of BRAF-mutated melanoma [64, 65]. Our group successfully rendered primary human NK cells resistant to the immunosuppressive transforming growth factor β1 (TGFβ) by targeted perturbation of the TGFBR2 gene using CRISPR/Cas9 editing [66]. Building on these observations, we were also able to demonstrate that disruption of TGFβ receptor signaling, both using small molecule inhibitors and genetic ablation, preserves NK cell function in a glioblastoma stem cell (GSC)-engrafted mouse model and prevents formation of a GSC-induced NK cell inhibitory phenotype [67].
Together these studies highlight how novel genetic engineering approaches can be employed to disarm the hostile tumor microenvironment by rendering CAR-modified immune cells resistant to some of the most notorious immunosuppressive mechanisms, a theme we project will continue to have important reverberations in the treatment of human cancers, particularly when targeting solid tumors.
Designing safe CAR-NK products for clinical use
While these new tools allow to introduce deliberate genetic interventions, opening up opportunities to extend the scope and potency of cellular immunotherapies, they pose a theoretical risk of non-specific off-target genetic lesions resulting in unintended adverse outcomes [68]. Hence, careful safety considerations are required, particularly when aiming to administer CRISPR/Cas9 gene-edited cell products in the clinic. Recent innovations, that include the transient delivery of high-fidelity Cas9 ribonucleoprotein complexes through electroporation, mitigate most of these undesired genomic alterations and powerful sequencing technologies [69,70,71] that are sensitive enough to detect even the rarest off-site lesions provide an additional safety margin for genetically engineered lymphocytes for clinical use.
Modulating CAR-NK metabolism
A growing body of work has established the crucial role of rapid metabolic reconfigurations required for NK cells to effectively mount anti-tumor responses [72, 73]. Targeting of immune-metabolic pathways, hence, has emerged as an intriguing concept to render NK cells more metabolically robust and preserve their functional capacity. In NK cells, IFNγ production has been shown to be dependent on glucose-driven oxidative phosphorylation [74]. Once activated, NK cells undergo substantial metabolic changes, thereby increasing the rates of glycolysis and oxidative phosphorylation (OXPHOS) to support the energy requirements to mount effective immune responses [75,76,77]. NK cell metabolic requirements are highly context-specific; hence, regulatory pathways altering the metabolic machinery and configuration need to be meticulously orchestrated to meet the dynamic energy and biosynthesis demands. Mammalian target of rapamycin complex 1 (mTORC1) plays a key role in regulating NK cell metabolism and facilitates both early NK cell development as well as activation-induced expression of nutrient transporters and glycolytic enzymes to support functional responses in mature NK cells. Indeed, our group and others have demonstrated that disruption of CISH signaling metabolically reprograms CAR-NK cells to maintain elevated mTORC1 and MYC activity, promoting in vivo persistence and enhanced anti-tumor activity [61, 78, 79].
At the transcriptional level, sterol regulatory element-binding protein (SREBP) and MYC have been identified as the two central regulatory elements orchestrating the metabolic adaptations which enable activated NK cells to effectively mount immune responses. MYC signaling serves to adapt the metabolic machinery to the specific requirements of NK cells by upregulating the expression of glucose transporters and glycolytic enzymes as well as increasing mitogenesis. Intracellular MYC levels are extremely short lived and tightly regulated by the dynamic interplay of MYC protein biosynthesis and proteasomal degradation. One mechanism by which intracellular MYC protein levels are maintained in check in NK cells is by glycogen synthase kinase 3 (GSK3)-mediated phosphorylation with subsequent ubiquitination and proteasomal degradation. In patients with AML, GSK3 expression has been shown to be upregulated and linked to impaired NK cell cytotoxicity that can be restored by genetic or pharmacologic GSK repression [80]. In line with these observations, pharmacologic inhibition of GSK3 has been found to stabilize MYC levels in NK cells, thereby increasing their anti-tumor potency [81].
Maintaining NK cell mitochondrial fitness for memory formation
Deriving implications from the dynamic interplay of metabolic programs that underpin immune cell differentiation and functional responses has become an imperative to guide future genetic engineering approaches for building the next generation of CAR-NK immunotherapies. NK cells have traditionally been ascribed to the innate immune system. However, recent studies have increasingly challenged this paradigm as a result of the discovery of cytokine-induced memory-like phenotypes [82]. Todd A. Fehninger and his group translated this approach to the clinic and demonstrated that preactivation of isolated NK cells with IL-12, IL-15 and IL-18 robustly induced formation of a memory-like NK cell phenotype with enhanced effector function against myeloid leukemic blasts [83, 84]. Building on their observations, the authors enrolled patients with AML in a phase 1 clinical trial (NCT01898793) evaluating the concept of interleukin-preactivated NK cells, and observed clinical responses in five out of nine (56%) evaluable patients including four complete remissions (44%). Transferring the knowledge acquired to CAR-engineered NK cells, the same group recently reported on the successful application of CD19-CAR-modified memory-like NK cells against a xenograft lymphoma mouse model [85]. Several groups are now investigating and analyzing the immunometabolic signatures underpinning this distinct memory-like NK phenotype [73]. Mitochondrial fitness, in particular, has come to the center of attention with growing evidence linking mitochondrial dysfunction to impaired NK cell immune responses, and strategies to promote mitochondrial health to improve NK cell function are already under way [34, 86, 87].
CAR-NK immunotherapy in solid tumors
Although cellular immunotherapy has opened promising avenues of treatment for hematologic malignancies, these successes have so far not been successfully transferred to the treatment of solid tumors. Besides the intrinsic challenges of CAR-NK immunotherapy such as limited in vivo persistence and functional exhaustion, solid tumors pose a unique set of obstacles for cell-based treatment strategies routed in their complex clonal heterogeneity, shared antigen expression with healthy tissues, and the inherently hostile immunosuppressive microenvironment. Furthermore, solid tumors have evolved mechanisms to downregulate activating NK cell ligands on their surface and can effectively impair trafficking of immune cells into tumor beds. Despite these challenges, there has been a growing interest in combatting solid tumors using cellular therapeutics in multiple, currently ongoing, early phase studies targeting a diverse range of antigens including HER2 (glioblastoma; NCT03383978), PSMA (prostate cancer; NCT03692663), mesothelin (ovarian cancer; NCT03692637), MUC1 (advanced solid tumors; NCT02839954), NKG2D (advanced solid tumors; NCT03415100) and ROBO1 (advanced solid tumors; NCT03940820 and pancreatic cancer; NCT03941457).
Gene engineering for improved CAR-NK trafficking
To achieve clinically meaningful anti-tumor responses and enable the widespread adoption of engineered cellular therapeutics for the treatment of solid tumors, the next generation of CAR-NK therapies will need to be engineered for enhanced migration into tumor beds and improved ability to penetrate the barriers imposed by solid tumors. Several groups are now working on genetic modifications of NK chemokine receptors to allow more efficient NK homing into tumor sites. In renal cell carcinoma, NK cells modified to express supraphysiological levels of CXCR2 exhibited improved trafficking to sites of disease manifestation [88]. Similarly, forced expression of chemokine receptor 4 (CXCR4) on EGFRvIII-directed CAR-NK cells led to improved survival in an animal model of glioblastoma [89]. Yu Yugan Ng and colleagues recently tested ectopic expression of CXCR1 in a CAR targeting NKG2D ligands, and were able to demonstrate improved immune cell infiltration and augmented anti-tumor efficacy in a mouse model of peritoneal ovarian cancer [90].
Whereas many hematologic malignancies are inherently mono- or oligoclonal diseases, solid tumors are characterized by complex clonal landscapes with a high degree of heterogeneity. Single antigen-directed cellular therapies face significant obstacles combatting heterogeneous tumors driven by multiple co-evolving clones, each with its distinct immune phenotype and antigen expression pattern. Cell-based therapies applied to combat solid tumors must also disentangle the intricacies of shared antigen expression between tumor and healthy tissues to avoid inadvertent on-target, off-tumor toxicities.
Dual and multi-specific targeting has emerged as a promising concept to mitigate the challenges imposed by heterogeneous and shared antigen expression. In the effort to devise therapies against highly refractory diseases such as glioblastoma, NK-92-derived natural killer cells have been equipped with a dual-specific CAR construct with specificity towards both wild type EGFR and EGFRvIII. When injected into two different xenograft models of GBM, the dual-targeting CAR-NK cells effectively negated tumor growth and improved survival independent of the EGRF mutation status [91]. In early 2021, Jeffrey Miller’s group presented their work on multi-specific off-the-shelf B7-H3-targeted iPSC-derived CAR-NK cells which can recognize a broad range of solid tumors. B7-H3 (CD276), a member of the B7 protein superfamily, is upregulated on a wide range of tumors, particularly in the metastatic setting and has been linked to poor prognosis [92]. In their study, the authors were able to detect strong cytotoxicity signals in vitro against ovarian, prostate, bone, and breast cancer cell lines. Despite its conspicuous appeal as pan-cancer antigen, recent reports of potential on-target off-tumor toxicities have surfaced which led to the temporary halt of an ongoing phase 1 clinical study evaluating a bispecific T cell engager targeting B7-H3 and CD3 [93]. Efforts to advance B7-H3-directed therapies into the clinic, hence, will need to proceed with prudence and carefully monitor any signals of emerging safety concerns.
Adoptive CAR-NK immunotherapy for the treatment of viral pathogens including COVID-19
Although most applications of CAR immunotherapy have thus far focused on addressing cancer, adoptive cell-based therapies have gained much notice as a powerful route to treat life-threatening treatment-refractory viral infections, particularly in highly vulnerable immunocompromised patient populations including those with primary immunodeficiencies and allogeneic stem cell recipients [94].
Virus-specific T cells (VST), both donor-derived and from third-party cell banks, continue to be explored in the treatment of several viral pathogens including cytomegalovirus, Epstein–Barr virus, adenovirus, HHV6, BK virus and JC virus [94,95,96,97]. Against the backdrop of the ongoing global COVID-19 pandemic, many groups have rushed to translate the learnings from virus-specific T cells to address SARS-CoV-2 infections using novel cellular based approaches. While the share of vaccinated people has in recent months risen markedly in most developed countries, there still is a strong unmet need for effective treatments for high-risk and immunocompromised patients who might not achieve sustained neutralizing antibody responses. Legitimate concerns about the eventual emergence of immune escape mutants including those which can evade current vaccination and treatment efforts, remain, and thus uphold the interest in adoptive cell-based approaches for the treatment of COVID-19.
Our group successfully expanded SARS-CoV-2-directed cytotoxic T cells (CTL) from the peripheral blood of recovered donors [98] and is currently leading a phase 1/2 trial to assess the feasibility, safety and efficacy of SARS-CoV-2 virus-specific T cells (VST) in patients with mild to moderate COVID-19 disease (NCT047422595). With increasing evidence supporting the use of corticosteroids to improve outcomes for patients with COVID-19, we furthermore propose a novel engineering strategy delete the gene encoding for the endogenous glucocorticoid receptor (NR3C1) of these donor-derived SARS-CoV-2-specific VSTs using CRISPR/Cas9 gene editing, thereby rendering the lymphocytes resistant to the immunosuppressive potential of corticosteroids [98].
Aside from isolating and expanding SARS-CoV-2-directed cytotoxic lymphocytes from recovered donors, several other groups have been exploring ways to specifically modify natural killer cells to recognize and kill SARS-CoV-2-infected host cells. In 2020, a group in mainland China evaluated a novel dual NKG2D-ACE2-engineered cord-blood-derived CAR-NK cell candidate in its ability to combat COVID-19 and is testing this approach in an ongoing clinical phase 1/2 trial (NCT04324996). Genetically equipped with constitutive Il-15 cytokine support and targeting both the highly conserved S protein of SARS-CoV-2 through expression of its natural receptor ACE2, as well as NKG2D ligands on infected cells by encoding for the multi-specific activating receptor NKG2D, their investigational CAR construct may be an interesting approach for the treatment of high-risk COVID-19 patients. Aiming to negate cytokine release syndrome and potential neurotoxicity, the construct has been designed to secrete a neutralizing scFv fragment against GM-CSF which has been implicated in the onset and progression of CRS and ICANS [99]. Very recently, Dongfang Liu’s group reported on the generation of NK-92 cell-line-derived CAR-NK cells targeting SARS-CoV-2 and its D614G mutant. Using the scFv fragment from SARS-CoV-2 neutralizing antibody S309, the authors were able to demonstrate improved in vitro cytotoxic activity against pseudotyped SARS-CoV-2 viral particles and pseudo-SARS-CoV-2 infected target cells compared to an earlier construct the group had previously reported on [100, 101].
In summary, these early reports on CAR-modified natural killer cells targeting SARS-CoV-2 illustrate the broad applicability and immense potential of CAR-NK immunotherapy even beyond cancer, and foreshadow an era of universal off-the-shelf immune cell-based strategies in the treatment of other severe and life-threatening diseases.
Stitching it all together—CAR-NK immunotherapy coming of age
Despite recent advances, fundamental questions regarding the clinical administration CAR-NK cell immunotherapy remain. These include optimal patient selection, as well as ideal therapeutic sequencing of CAR-NK immunotherapy in the context of other established treatment regimens including autologous and allogeneic stem cell transplantation for hematologic malignancies. Future studies will need to address how to best select high-risk patients for CAR-NK immunotherapy by identifying prognostic biomarkers and assessing the optimal sequencing of CAR-redirected NK therapies to feed into established treatment algorithms.
While the recent report of the first successful clinical application of CAR-NK-based cellular therapy against high-risk lymphoid malignancies represents a major milestone in the field of cancer immunotherapy [24], it only marks the beginning of an exponential wave of cellular engineering innovations in the upcoming years. Going forward, we project that while conventional, non-antigen-specific cell-based therapies in the form of autologous and allogeneic stem cell transplant will continue to play an important role in the management of high-risk patients with hematologic malignancies for the foreseeable future, it is likely that antigen-specific CAR-based therapies will, over time, move up to earlier lines of therapy. Allogeneic off-the-shelf CAR-NK cell-based therapies are especially poised to supplant long-held treatment paradigms due to their broad applicability in the allogeneic setting and their tremendous cost–benefit compared to autologous cell products. For solid tumors, first interim reports of the currently ongoing clinical trials are highly anticipated and will allow for a more robust perspective on how fast cellular immunotherapy can conquer this area of high unmet medical need. Encouragingly, research activity has been spurred in recent years, and many promising CAR-NK cell candidates leveraging highly innovative next-generation genetic engineering approaches are underway to address the unique challenges of solid tumors and expected to advance to clinical testing in the near term. Underpinning these developments is the ever-growing understanding of fundamental immune cell biology, increasingly driven by powerful multi-omics and high-dimensional single cell analyses, which in many ways continue to lay the groundwork for a burst of innovations in the near future. Turbocharged with the powerful advances in genetic engineering capabilities, these insights are poised to spur further therapeutic innovations and, over time, radically alter long-held treatment paradigms and dramatically improve clinical outcomes to the benefit of generations of patients to come.
Abbreviations
- A2AR:
-
A2A adenosine receptor
- ACE2:
-
Angiotensin-converting enzyme 2
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- ALCL:
-
Anaplastic large cell lymphoma
- AML:
-
Acute myeloid leukemia
- BCMA:
-
B-cell maturation antigen
- B-NHL:
-
B-cell non-Hodgkin lymphoma
- Breg:
-
Regulatory B cell
- CAR-NK:
-
Chimeric antigen receptor-modified natural killer cell
- CISH:
-
Cytokine-inducible SH2-containing protein
- COVID-19:
-
Coronavirus disease 2019
- CR:
-
Complete remission
- CRISPaint:
-
CRISPR-assisted insertion tagging
- CRISPR/Cas9:
-
Clustered regularly interspaced short palindromic repeats/Caspase 9
- CRS:
-
Cytokine release syndrome
- CTL:
-
Cytotoxic T cells
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCR1/2/4:
-
Chemokine receptor 1 (CXCR1); chemokine receptor 2 (CXCR2); chemokine receptor 4 (CXCR4)
- DAP10:
-
DNAX-activating protein 10
- DAP12:
-
DNAX-activating protein 12
- EGFR:
-
Epidermal growth factor receptor
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- EMCN:
-
Endomucin
- FDA:
-
U.S. Food and Drug Administration
- FLT3:
-
Fms-like tyrosine kinase 3
- GBM:
-
Glioblastoma
- GSC:
-
Glioblastoma stem cell
- GSK3:
-
Glycogen synthase kinase 3
- GvHD:
-
Graft versus host disease
- HER2:
-
Receptor tyrosine-protein kinase erbB-2
- HHV6:
-
Human herpesvirus 6
- HLA:
-
Human leukocyte antigen
- HSC:
-
Hematopoietic stem cell
- ICANS:
-
Immune cell-associated neurologic toxicity
- iCasp9:
-
Inducible caspase 9
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFNγ:
-
Interferon gamma
- Il-12/15/18:
-
Interleukin 12/15/18
- iPSC:
-
Induced pluripotent stem cells
- JC virus:
-
Human polyomavirus 2
- KLRG1:
-
Killer cell lectin-like receptor subfamily G member 1
- LAG-3:
-
Lymphocyte-activation gene 3
- mAb:
-
Monoclonal antibody
- MDACC:
-
The University of Texas MD Anderson Cancer Center
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC-1:
-
Major histocompatibility complex 1
- mTORC1:
-
Mammalian target of rapamycin complex 1
- MUC1:
-
Mucin 1
- NIH:
-
National Institutes of Health
- NK:
-
Natural killer cells
- OXPHOS:
-
Oxidative phosphorylation
- PBMC:
-
Peripheral blood mononuclear cells
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- PGE2:
-
Prostaglandin E2
- PSCA:
-
Prostate stem cell antigen
- PSMA:
-
Prostate-specific membrane antigen
- R/R:
-
Relapsed or refractory
- ROBO1:
-
Roundabout homolog 1
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- scFv:
-
Single-chain variable fragment
- SREBP:
-
Sterol regulatory element-binding protein
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TAMs:
-
Tumor-associated macrophages
- TCR:
-
T-cell receptor
- TGFβ:
-
Transforming growth factor beta 1
- TGFBR2:
-
Transforming growth factor beta receptor 2
- TIGIT:
-
T-cell immunoreceptor with Ig and ITIM domains
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing-3
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- T-NHL:
-
T-cell non-Hodgkin lymphoma
- Treg:
-
Regulatory T cell
- VST:
-
Virus-specific T cell
- WT:
-
Wild type
References
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–37.
Ulrich Jaeger, Michael R. Bishop, Gilles Salles, Stephen J. Schuster, Richard T. Maziarz, Xia Han, Alexander Savchenko, Nathan Roscoe, Elena Orlando, Dawson Knoblock, Ranjan Tiwari, Lida Bubuteishvili Pacaud and Paolo Corradini. Myc Expression and Tumor-Infiltrating T Cells Are Associated with Response in Patients (Pts) with Relapsed/Refractory Diffuse Large B-Cell Lymphoma (r/r DLBCL) Treated with Tisagenlecleucel in the Juliet Trial. Presented at: 2020 ASH Annual Meeting, December 5–8; virtual; Session: 626 Abstract 1194. 2020. https://ash.confex.com/ash/2020/webprogram/Paper137045.html.
Caron Jacobson, Frederick L. Locke, Armin Ghobadi, David B. Miklos, Lazaros J. Lekakis, Olalekan O. Oluwole, Yi Lin, Ira Braunschweig, Brian T. Hill, John M. Timmerman, Abhinav Deol, Patrick M. Reagan, Patrick J. Stiff, Ian W. Flinn, Umar Farooq, Andre H. Goy, Peter A. McSweeney, Javier Munoz, Tanya Siddiqi, John M. Rossi, Adrian Bot, Lianqing Zheng, Remus Vezan, Zahid Bashir, Jenny J. Kim, Rong Chu and Sattva S. Neelapu. Long-term survival and gradual recovery of B cells in patients with refractory large B cell lymphoma treated with axicabtagene ciloleucel (Axi-Cel). Presented at: 2020 ASH annual Meeting, December 5–8; virtual; Session: 626 Abstract 1187. 2020. https://ash.confex.com/ash/2020/webprogram/Paper134362.html.
Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021. https://doi.org/10.1038/s41591-021-01436-0.
Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021;18(6):379–93.
Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–56.
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–84.
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20(1):70–5.
Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129(12):2935–44.
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci. 2009;106(9):3360.
Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:283.
Rafei H, Daher M, Rezvani K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br J Haematol. 2021;193(2):216–30.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135(6):399–410.
Strati P, Bachanova V, Goodman A, Pagel JM, Castro JE, Griffis K, et al. Preliminary results of a phase I trial of FT516, an off-the-shelf natural killer (NK) cell therapy derived from a clonal master induced pluripotent stem cell (iPSC) line expressing high-affinity, non-cleavable CD16 (hnCD16), in patients (pts) with relapsed/refractory (R/R) B-cell lymphoma (BCL). J Clin Oncol. 2021;39(15_suppl):7541.
Bachier C, Borthakur G, Hosing C, Blum W, Rotta M, Ojeras P, et al. A Phase 1 study of NKX101, an allogeneic CAR natural killer (NK) cell therapy, in subjects with relapsed/refractory (R/R) acute myeloid leukemia (AML) or higher-risk myelodysplastic syndrome (MDS). Blood. 2020;136(Supplement 1):42–3.
Goodridge JP, Mahmood S, Zhu H, Gaidarova S, Blum R, Bjordahl R, et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood. 2019;134(Supplement_1):301.
Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. 2016;7(35):56219–32.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8(68):112783–96.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64–78.
Fleischer LC, Spencer HT, Raikar SS. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J Hematol Oncol. 2019;12(1):141.
Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30+ malignancies. Clin Cancer Res. 2021;27(13):3744.
Cummins KD, Gill S. Chimeric antigen receptor T-cell therapy for acute myeloid leukemia: how close to reality? Haematologica. 2019;104(7):1302.
Kerbauy LN, Ang S, Liu E, Banerjee PP, Wu Y, Shaim H, et al. Cord blood NK cells engineered to express a humanized CD123-targeted chimeric antigen receptor (CAR) and IL-15 as off-the-shelf therapy for acute myeloid leukemia. Blood. 2017;130(Supplement 1):4453.
Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28(10):897–913.
Salman H, Pinz KG, Wada M, Shuai X, Yan LE, Petrov JC, et al. Preclinical targeting of human acute myeloid leukemia using CD4-specific chimeric antigen receptor (CAR) T cells and NK cells. J Cancer. 2019;10(18):4408.
Naeimi Kararoudi M, Likhite S, Elmas E, Schwartz M, Sorathia K, Yamamoto K, et al. CD33 targeting primary CAR-NK cells generated By CRISPR mediated gene insertion show enhanced anti-aml activity. Blood. 2020;136(Supplement 1):3.
Gonzalez A, Roguev A, Frankel NW, Garrison BS, Lee D, Gainer M, et al. Abstract LB028: Development of logic-gated CAR-NK cells to reduce target-mediated healthy tissue toxicities. Can Res. 2021;81(13 Supplement):LB028.
Gonzalez A, Roguev A, Frankel NW, Garrison BS, Lee D, Gainer M, et al. Abstract LB028: Development of logic-gated CAR-NK cells to reduce target-mediated healthy tissue toxicities. Cancer Res. 2021;81(13 Supplement):LB028-LB.
Brian S. Garrison, Han Deng, Gozde Yucel, Nicholas W. Frankel, Marcela Ayala Guzman, Russell Gordley, Michelle Hung, Derrick Lee, Marcus Gainer, Kathryn Loving, Jenny Chien, Tiffany Pan, Wesley Gorman, Travis Wood, Wilson Wong, Philip Lee, Tim Lu, Gary Lee. Precise Targeting of AML with First-inClass OR / NOT Logic-Gated Gene Circuits in CAR-NK Cells. In: Presented at the 24th ASGCT Annual Meeting Abstract 77. 2021; 29 No 4S1. https://www.cell.com/action/showPdf?pii=S1525-0016%2821%2900206-9.
Campbell KS, Cohen AD, Pazina T. Mechanisms of NK cell activation and clinical activity of the therapeutic SLAMF7 antibody, elotuzumab in multiple myeloma. Front Immunol. 2018. https://doi.org/10.3389/fimmu.2018.02551.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2):297–310.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Elena Maroto-Martín, Jessica Encinas, Almudena García-Ortiz, Rafael Alonso, Alejandra Leivas, Mari Liz Paciello, Vanesa Garrido, Teresa Cedena, Laura Ugalde, Daniel J. Powell Jr., Paula Río, Joaquín Martinez-López, Antonio Valeri. NKG2D and BCMA-CAR NK cells efficiently eliminate multiple myeloma cells. A comprehensive comparision between two clinically relevant CARs. EHA Library. 06/15/19; 266826; PS1209. https://library.ehaweb.org/eha/2019/24th/266826/elena.maroto.martn.nkg2d.and.bcma-car.nk.cells.efficiently.eliminate.multiple.html.
Leivas A, Rio P, Mateos R, Paciello ML, Garcia-Ortiz A, Fernandez L, et al. NKG2D-CAR transduced primary natural killer cells efficiently target multiple myeloma cells. Blood. 2018;132(Supplement 1):590.
Martín EM, Encinas J, García-Ortiz A, Ugalde L, Fernández RA, Leivas A, et al. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019;19(10):e24–5.
Goodridge JP, Bjordahl R, Mahmood S, Reiser J, Gaidarova S, Blum R, et al. Abstract 1550: FT576 path to first-of-kind clinical trial: translation of a versatile multi-antigen specific off-the-shelf NK cell for treatment of multiple myeloma. Can Res. 2021;81(13 Supplement):1550.
Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. 2019;8(5):e1049.
Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003;4(6):557–64.
Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391(6668):703–7.
Nakajima H, Colonna M. 2B4: an NK cell activating receptor with unique specificity and signal transduction mechanism. Hum Immunol. 2000;61(1):39–43.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23(2):181-92.e5.
Pedroza-Pacheco I, Madrigal A, Saudemont A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol. 2013;10(3):222–9.
Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070–6.
Judge SJ, Murphy WJ, Canter RJ. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol. 2020;10:49.
Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Investig. 2019;129(5):2094–106.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Investig. 2018;128(10):4654–68.
Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 Is expressed by activated mouse NK cells and inhibits NK cell IFN-γ production in response to mature dendritic cells. J Immunol. 2014;192(9):4184–91.
Young A, Ngiow Shin F, Barkauskas Deborah S, Sult E, Hay C, Blake Stephen J, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. 2016;30(3):391–403.
Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Can Res. 2018;78(4):1003.
Daher M, Basar R, Shaim H, Gokdemir E, Uprety N, Kontoyiannis A, et al. The TGF-β/SMAD signaling pathway as a mediator of NK cell dysfunction and immune evasion in myelodysplastic syndrome. Blood. 2017;130(Supplement 1):53.
Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. 2021. https://doi.org/10.1172/JCI142116.
Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids. 2015;4(11):e264.
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187–97.
Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14(6):607–14.
Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, et al. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80.
O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. 2019;19(5):282–90.
Choi C, Finlay DK. Optimising NK cell metabolism to increase the efficacy of cancer immunotherapy. Stem Cell Res Ther. 2021;12(1):320.
Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol. 2015;194(4):1954.
Gardiner CM. NK cell metabolism. J Leukoc Biol. 2019;105(6):1235–42.
Kobayashi T, Mattarollo SR. Natural killer cell metabolism. Mol Immunol. 2019;115:3–11.
Poznanski SM, Ashkar AA. What defines NK cell functional fate: phenotype or metabolism? Front Immunol. 2019;10:1414.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27(2):224-37.e6.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, et al. CIS is a potent checkpoint in NK cell–mediated tumor immunity. Nat Immunol. 2016;17(7):816–24.
Parameswaran R, Ramakrishnan P, Moreton SA, Xia Z, Hou Y, Lee DA, et al. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat Commun. 2016;7(1):11154.
Cichocki F, Valamehr B, Bjordahl R, Zhang B, Rezner B, Rogers P, et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017;77(20):5664–75.
Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci. 2009;106(6):1915–9.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18–preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–65.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136(20):2308–18.
Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. 2019;20(12):1656–67.
Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M, et al. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2020-002044.
Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer. 2017;5(1):73.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. Journal of immunotherapy (Hagerstown, Md: 1997). 2015;38(5):197.
Ng YY, Tay JC, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncol. 2020;16:75–85.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-Engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5(1):11483.
Bjordahl R, Goulding J, Chu H-Y, Cichocki F, Zorko N, Davis Z, et al. Abstract 1539: development of off-the-shelf B7H3 chimeric antigen receptor NK cell therapeutic with broad applicability across many solid tumors. Can Res. 2021;81(13 Supplement):1539.
Zhang Z, Jiang C, Liu Z, Yang M, Tang X, Wang Y, et al. B7-H3-targeted CAR-T cells exhibit potent antitumor effects on hematologic and solid tumors. Mol Ther Oncol. 2020;17:180–9.
Keller MD, Bollard CM. Virus-specific T-cell therapies for patients with primary immune deficiency. Blood. 2020;135(9):620–8.
Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, epstein-barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2017;35(31):3547–57.
Muftuoglu M, Olson A, Marin D, Ahmed S, Mulanovich V, Tummala S, et al. Allogeneic BK virus-specific T cells for progressive multifocal leukoencephalopathy. N Engl J Med. 2018;379(15):1443–51.
Olson A, Lin R, Marin D, Rafei H, Bdaiwi MH, Thall PF, et al. Third-party BK virus-specific cytotoxic T lymphocyte therapy for hemorrhagic cystitis following allotransplantation. J Clin Oncol. 2019;134:3596.
Basar R, Uprety N, Ensley E, Daher M, Klein K, Martinez F, et al. Generation of glucocorticoid-resistant SARS-CoV-2 T cells for adoptive cell therapy. Cell Rep. 2021. https://doi.org/10.1016/j.celrep.2021.109432.
Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709.
Ma MT, Badeti S, Chen C-H, Kim J, Choudhary A, Honnen B, Reichman C, Calianese D, Pinter A, Jiang Q, Shi L, Zhou R, Xu H, Li Q, Gause W and Liu D (2021) CAR-NK Cells Effectively Target SARS-CoV-2-Spike-Expressing Cell Lines In Vitro. Front. Immunol. 12:652223. https://doi.org/10.3389/fimmu.2021.652223.
Efficacy of Targeting SARS-CoV-2 by CAR-NK CellsMinh Ma, Saiaditya Badeti, Ke Geng, Dongfang LiubioRxiv 2020.08.11.247320. https://doi.org/10.1101/2020.08.11.247320.
Acknowledgements
A.B. received support from the German Research Foundation as Walter Benjamin Postdoctoral Fellow. This work was made possible by the generous support of Ann and Clarence Cazalot, by a generous donation from Lyda Hill Philanthropies, and The University of Texas MD Anderson Cancer Center Moon Shots Program; by Grants from CPRIT (RP160693), by a Stand Up To Cancer Dream Team Research Grant (Grant number: SU2C-AACR-DT-29-19), by Grants (1 R01 CA211044-01, 5 P01CA148600-03, and P50CA100632-16) from the National Institutes of Health (NIH), the Specialized Program of Research Excellence (SPORE) in Brain Cancer Grant (P50CA127001), and by a Grant (CA016672) to the MD Anderson Cancer Center from the NIH. The SU2C research grant is administered by the American Association for Cancer Research, the scientific partner of SU2C.
Author information
Authors and Affiliations
Contributions
AB and KR wrote and approved the review article.
Corresponding author
Ethics declarations
Conflict of interest
K.R., and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to CAR-NK cell research. MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MDACC’s conduct of any other ongoing or future research related to this relationship. K.R., and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Affimed GmbH. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson is implementing an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’s conduct of any other ongoing or future research related to this relationship. K.R. participates on the Scientific Advisory Board for GemoAb, AvengeBio, Virogin, GSK, Caribou and Bayer.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Biederstädt, A., Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int J Hematol 114, 554–571 (2021). https://doi.org/10.1007/s12185-021-03209-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-021-03209-4