
Abstract
β-Phosphoglucomutase (βPGM) is a magnesium-dependent phosphoryl transfer enzyme that catalyses the reversible isomerisation of β-glucose 1-phosphate and glucose 6-phosphate, via two phosphoryl transfer steps and a β-glucose 1,6-bisphosphate intermediate. Substrate-free βPGM is an essential component of the catalytic cycle and an understanding of its dynamics would present significant insights into βPGM functionality, and enzyme catalysed phosphoryl transfer in general. Previously, 30 residues around the active site of substrate-free βPGMWT were identified as undergoing extensive millisecond dynamics and were unassignable. Here we report 1H, 15N and 13C backbone resonance assignments of the P146A variant (βPGMP146A) in its substrate-free form, where the K145–A146 peptide bond adopts a trans conformation in contrast to all crystal structures of βPGMWT, where the K145–P146 peptide bond is cis. In βPGMP146A millisecond dynamics are suppressed for all but 17 residues, allowing 92% of backbone resonances to be assigned. Secondary structure predictions using TALOS-N reflect βPGM crystal structures, and a chemical shift comparison between substrate-free βPGMP146A and βPGMWT confirms that the solution conformations are very similar, except for the D137–A147 loop. Hence, the isomerisation state of the 145–146 peptide bond has little effect on structure but the cis conformation triggers millisecond dynamics in the hinge (V12–T16), the nucleophile (D8) and residues that coordinate the transferring phosphate group (D8 and S114–S116), and the D137–A147 loop (V141–A142 and K145). These millisecond dynamics occur in addition to those for residues involved in coordinating the catalytic MgII ion and the L44–L53 loop responsible for substrate discrimination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biological context
β-Phosphoglucomutase (βPGM, EC 5.4.2.6) from Lactococcus lactis is a magnesium-dependent phosphoryl transfer enzyme of the haloacid dehalogenase superfamily (Lahiri et al. 2002a; Allen and Dunaway-Mariano 2004; Dai et al. 2009). In the catabolism of maltose and trehalose, βPGM catalyses the reversible isomerisation of β-glucose 1-phosphate (βG1P) and glucose 6-phosphate (G6P). During catalysis, βG1P binds to phosphorylated βPGM (βPGMP, phosphorylated on residue D8) forming β-glucose 1,6-bisphosphate (βG16BP), which is released to solution. Subsequent rebinding of βG16BP in the alternate orientation to non-phosphorylated, substrate-free βPGM results in dephosphorylation of βG16BP, with the formation of G6P (which enters glycolysis) and the regeneration of βPGMP (Zhang et al. 2005; Dai et al. 2006). The βPGM gene (pgmB) is located on the trehalose operon and is induced by maltose or trehalose in the growth medium but is repressed by the presence of glucose or lactose (Qian et al. 1994, 1997). A βPGM knockout mutant strain of L. lactis shows impaired growth when maltose is used as the only carbon source, coupled with an intracellular accumulation of trehalose 6-phosphate and polysaccharide molecules composed of α-1,4-linked glucose units (Levander et al. 2001). Such a perturbation of the metabolic flux highlights the crucial role that βPGM plays in mediating the efficient utilisation of carbohydrate species in L. lactis metabolism.
Wild-type βPGM (βPGMWT) together with a series of variants have been studied extensively using kinetic experiments (Zhang et al. 2005; Dai et al. 2006, 2009; Goličnik et al. 2009), X-ray crystallography (Lahiri et al. 2002a, b, 2003; Tremblay et al. 2005; Baxter et al. 2010; Griffin et al. 2012; Jin et al. 2014; Johnson et al. 2018), NMR spectroscopy (Baxter et al. 2006, 2008, 2009, 2010; Griffin et al. 2012; Jin et al. 2014; Johnson et al. 2018) and density functional theory approaches (Webster 2004; Marcos et al. 2010, Elsässer et al. 2012; Barrozo et al. 2018) and it is considered as an archetypal system for enzyme catalysed phosphoryl transfer reactions. Structural analysis coupled with metal-fluoride ground state and transition state analogue (TSA) complexes have allowed the atomic resolution description of several discrete species found in the catalytic cycle i.e. substrate-free βPGMWT (PDB: 2WHE; Baxter et al. 2010), a ground state βPGMPWT analogue (βPGMWT:BeF3 complex; PDB: 2WFA; Griffin et al. 2012), two ground state βPGMPWT:G6P complexes (βPGMWT:BeF3:G6P complexes; PDB: 2WF8; PDB: 2WF9; Griffin et al. 2012), two βPGMD10N:βG16BP complexes (PDB: 5OK1; PDB: 5OK0; Johnson et al. 2018), a βPGMPWT:G6P TSA complex (βPGMWT:MgF3:G6P TSA complex; PDB: 2WF5; Baxter et al. 2010) and a βPGMPWT:βG1P TSA complex (βPGMWT:MgF3:βG1CP TSA complex; PDB: 4C4R; Jin et al. 2014). The enzyme active site is located in the cleft formed between the α/β core domain (M1–D15 and S88–K216) and the α-helical cap domain (T16–V87). During catalysis, domain reorientation through hinge residue (D15–T16 and V87–S88) rearrangement results in closure and opening of the active site cleft facilitating substrate binding and product release. Two phosphate group binding sites are present, one in a proximal site adjacent to the carboxylate nucleophile (residue D8) (Lahiri et al. 2002a) and the catalytic MgII ion (Lahiri et al. 2002a), and the other in a distal site located ~ 8 Å away in the closed enzyme (Lahiri et al. 2003). The carboxylate group of the assigned general acid–base (residue D10) (Dai et al. 2009) populates two orientations depending on the degree of active site closure. In the structures of substrate-free βPGMWT and the βPGMPWT analogue, the active site cleft is open and the D10 carboxylate group is not engaged in the active site, whereas in the closed βPGMPWT:G6P, βPGMD10N:βG16BP, βPGMPWT:G6P TSA and βPGMPWT:βG1P TSA complexes, the carboxylate group is positioned to facilitate general acid–base catalysis promoting phosphoryl transfer (Johnson et al. 2018). Key roles for several residue segments in the active site have been identified including, coordination of the transferring phosphate group in the proximal site (V9, D10, S114, A115 and K145) (Lahiri et al. 2003), coordination of the phosphate group of the substrate in the distal site (R49, S116, K117 and N118) (Lahiri et al. 2003), substrate discrimination and binding (L44–L53) (Lahiri et al. 2004) and coordination of the catalytic MgII ion (D10, E169 and D170) (Lahiri et al. 2002a).
Previously, the solution behaviour of substrate-free βPGMWT was investigated by NMR spectroscopy and a backbone resonance assignment was determined (BMRB: 7235; Baxter et al. 2006). However, 30 residues (D8–T16, R38, L44–L53, S114–N118, V141–A142, K145 and S171–Q172) located primarily in the active site loops remained unassigned in the 1H–15N TROSY spectrum, most likely due to extensive conformational intermediate exchange dynamics occurring on the millisecond timescale, which results in broadening of the correlations beyond the limits of detection. Substrate-free βPGM is an essential component of the catalytic cycle and an understanding of the dynamics of key residue segments would present significant insights into βPGM functionality. Accordingly, a series of single site variants of βPGM was screened to establish whether any improvement in spectral quality could be obtained. Of the variants tested, the P146A variant of βPGM (βPGMP146A) reduced the intermediate exchange dynamics by the strongest extent and so was investigated further. On the basis of the conformational properties of alanine, βPGMP146A is expected to adopt a trans K145–A146 peptide bond as the dominant population. In contrast, all of the reported crystal structures described for βPGMWT, indicate a cis K145–P146 peptide bond within the D137–A147 loop. Consequently the isomerisation state of the 145–146 peptide bond presents a trigger for some of the intermediate exchange dynamics observed. Preliminary kinetics experiments using methods described previously (Johnson et al. 2018) indicate that βPGMP146A is active. Complete equilibration of 10 mM βG1P with G6P by 3 μM βPGMP146A was achieved in 1.5 h. Here, we report the 1HN, 15N, 13Cα, 13Cβ and 13C’ backbone resonance assignments of substrate-free βPGMP146A, including the resonances of many residues that were previously unassigned in substrate-free βPGMWT.
Methods and experiments
Protein expression and purification
Site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis Kit, Agilent Technologies) of the pgmB from Lactococcus lactis cloned in the pET-22b(+) expression plasmid was employed to generate βPGMP146A using primers with single-site base changes. Successful mutagenesis was confirmed by DNA sequencing. The plasmid was transformed into Escherichia coli strain BL21(DE3) cells (Stratagene) and 2H,15N,13C-labelled βPGMP146A (25 kDa) was expressed in defined isotopically labelled minimal media (Reed et al. 2003). The cells were grown at 37 °C with shaking until OD600nm = 0.6, at which point they were cooled to 25 °C and induced with isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. Cells were incubated for a further 18 h and were harvested by centrifugation at 10,000 rpm for 10 min. The cell pellet was resuspended in ice-cold standard working buffer (50 mM K+ HEPES pH 7.2, 5 mM MgCl2, 2 mM NaN3, 1 mM EDTA) supplemented with cOmplete™ protease inhibitor cocktail (Roche) (one tablet per 50 mL cell suspension). The cell suspension was lysed on ice using 6 cycles of sonication with pulsation for 20 s followed by 60 s cooling intervals. The cell lysate was then separated by ultracentrifugation at 20,000 rpm (Beckman Coulter Avanti centrifuge using rotor JA-20) for 35 min at 4 °C. The cleared cell lysate was filtered using a 0.22 µm syringe filter (Merck Millipore) and loaded onto a DEAE-Sepharose fast flow anion-exchange column connected to an ÄKTA purification system (GE Healthcare) that had been washed previously with 1 column volume of 6 M guanidine chloride, 1 column volume of 1 M NaOH and equilibrated with 5 column volumes of standard working buffer. Proteins bound to the DEAE-Sepharose column were eluted with a gradient of 0 to 50% standard working buffer containing 1 M NaCl. Fractions were checked for the presence of βPGMP146A by SDS-PAGE, pooled together and concentrated by Vivaspin (10 kDa MWCO, Sartorius). The protein sample was loaded onto a prepacked Hiload 26/60 Superdex 75 size-exclusion column connected to an ÄKTA purification system previously washed with 1 column volume of 1 M NaOH and equilibrated with 1.5 column volumes of standard working buffer containing 1 M NaCl. Fractions containing βPGMP146A were checked for purity by SDS-PAGE, pooled together and buffer exchanged into standard working buffer and concentrated to ~ 1.6 mM by Vivaspin (10 kDa MWCO) for storage as 1 mL aliquots at − 20 °C. No procedure was necessary to promote back exchange to amide protium atoms in perdeuterated βPGMP146A. Protein concentrations were estimated by absorbance at 280 nm (ε280 = 19940 M−1 cm−1). All reagents were of analytical grade and were purchased from Sigma-Aldrich (UK), except for the stable isotopically-labelled compounds 15NH4Cl (99%), 13C, 2H7-d-Glucose (U–13C6, 99%; 1,2,3,4,5,6,6-d7 97–98%) and 2H2O (99.8%), which were purchased from CortecNet (France) and used as received.
NMR experiments
The NMR experiments were acquired using samples loaded into 5-mm NMR tubes, which contained 1.2 mM 2H,15N,13C-labelled βPGMP146A in standard working buffer supplemented with 2H2O (10% v/v) for the deuterium lock and 1 mM trimethylsilyl propanoic acid (TSP) as a chemical shift reference. All experiments were recorded at 298 K using an 800 MHz Bruker Avance I spectrometer fitted with a 5-mm TXI probe equipped with z-axis gradients and running TopSpin software version 2.1. For the backbone 1H, 15N, 13C resonance assignment of substrate-free βPGMP146A, 2D 1H–15N TROSY and TROSY-based 3D HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, HN(CA)CO and HNCO spectra were acquired using standard Bruker pulse sequences. 1H chemical shifts were referenced relative to the internal TSP signal resonating at 0.0 ppm, whereas 15N and 13C chemical shifts were referenced indirectly using nuclei-specific gyromagnetic ratios.
Resonance assignments and data deposition
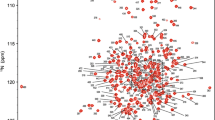
Backbone 1HN, 15N, 13Cα, 13Cβ and 13C’ chemical shifts were assigned for substrate-free βPGMP146A using standard triple resonance methodology (Gardner and Kay 1998). The processing of spectra and peak picking were performed using FELIX (Felix NMR, Inc.). Frequency matching of the backbone assignments was achieved using a simulated annealing algorithm employed by the “asstools” assignment program (Reed et al. 2003). The backbone 1HN, 15N, 13Cα, 13Cβ and 13C’ chemical shifts have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/) under the BMRB accession code 27920. Excluding the nine proline residues and the N-terminal methionine residue, 194 out of a possible 211 residues were assigned in the 1H–15N TROSY spectrum (Fig. 1). In total, 93.4% of all backbone resonances were assigned (91.9% of 1HN, 91.9% of 15N, 94.6% of 13Cα, 93.7% of 13Cβ and 94.6% of 13C′ nuclei).

1H–15N TROSY spectrum of 2H,15N,13C-labelled substrate-free βPGMP146A in 50 mM K+ HEPES pH 7.2, 5 mM MgCl2, 2 mM NaN3, 1 mM EDTA, 10% v/v 2H2O and 1 mM TSP recorded on an 800 MHz spectrometer at 298 K. a The full spectrum is shown together with b an expansion of the crowded region. The assignments of the backbone amide resonances are indicated by residue type and sequence number
There are 17 residues that remain unassigned in the 1H–15N TROSY spectrum of substrate-free βPGMP146A (L9, D10, G11, R38, L44, K45, G46, S48, R49, E50, D51, S52, L53, K117, N118, D170 and S171) compared with 30 residues in substrate-free βPGMWT (Fig. 2). From the crystal structures of substrate-free βPGMWT (PDB: 2WHE) and the βPGMWT:MgF3:G6P TSA complex (PDB: 2WF5), all of the unassigned residues in substrate-free βPGMP146A (except for R38) are situated within the active site and have significant roles in the catalytic cycle of the enzyme. Residues L9, D10 and G11 are key components of the proximal site, with D8 identified as the nucleophile, D10 assigned as the general acid–base and residues D10, D170 and S171 comprising the catalytic MgII ion binding site. Residues R49, K117 and N118 coordinate the phosphate group of the substrate in the distal binding site and the active site loop L44–L53 in the cap domain comprises part of a helix–loop–helix motif and is involved in substrate discrimination and binding. Due to the involvement of these residue segments in the catalytic cycle, it is likely that conformational exchange dynamics between two (or more) similarly populated forms is still occurring on the millisecond timescale in substrate-free βPGMP146A, resulting in the attenuation of 1H–15N TROSY correlations beyond the limits of detection. Increased solvent exposure of the amide group of R38 through perturbation of hydrogen bonding with neighbouring sidechain groups may be coupled with helix-fraying exchange behaviour within the second α-helix (D37–E42) and the first turn of the third α-helix (S48–D58) of the cap domain, resulting in a loss of the 1H–15N TROSY correlation.

Cartoon representation of the substrate-free βPGMWT crystal structure (PDB: 2WHE) highlighting the extent of backbone amide resonance assignments for substrate-free βPGMP146A and substrate-free βPGMWT. Assigned residues for βPGMP146A are coloured white (for loops and α-helices) and tan (for β-strands), with proline residues coloured green and residues that were unassigned in βPGMWT coloured cyan. Unassigned residues for βPGMP146A (L9, D10, G11, R38, L44, K45, G46, S48, R49, E50, D51, S52, L53, K117, N118, D170 and S171) are coloured purple and the 30 unassigned residues for βPGMWT are listed here for comparison (D8, L9, D10, G11, V12, I13, T14, D15, T16, R38, L44, K45, G46, V47, S48, R49, E50, D51, S52, L53, S114, A115, S116, K117, N118, V141, A142, K145, S171 and Q172). The location of the P146A mutation site is highlighted with a red backbone and the catalytic magnesium ion is shown as a green sphere
The secondary structure content and residue-specific random coil index order parameters (RCI-S2) of substrate-free βPGMP146A were predicted by uploading the backbone 1HN, 15N, 13Cα, 13Cβ and 13C’ chemical shifts to the TALOS-N webserver (Shen and Bax 2013). The predicted secondary structure for the solution conformation of substrate-free βPGMP146A compares well with the secondary structure present in the substrate-free βPGMWT crystal (PDB: 2WHE) (Fig. 3). In addition, residues with the highest values of RCI-S2 are located in well-defined secondary structure elements, whereas residues with the lowest values (having more random coil-like chemical shifts) correspond to loop regions in the crystal structure. Together, these data are in very good agreement, which indicates that the solution conformation is similar to the protein structure observed in the substrate-free βPGMWT crystal and provides confidence in the assignments of substrate-free βPGMP146A.

Predicted secondary structure content and residue-specific random coil index order parameters (RCI-S2) of substrate-free βPGMP146A obtained with the TALOS-N webserver (Shen and Bax 2013) using the backbone 1HN, 15N, 13Cα, 13Cβ and 13C′ chemical shifts. The secondary structure prediction is shown as red bars for α-helices and blue bars for β-strands, with the height of the bars representing the probability assigned by the software. As a comparison, the secondary structure observed in the substrate-free βPGMWT crystal (PDB: 2WHE) is shown at the top of the figure in the same colour representation. The predicted RCI-S2 values are shown as black circles
A chemical shift comparison between substrate-free βPGMP146A and substrate-free βPGMWT (BMRB: 7235; Baxter et al. 2006) also supports the conclusion that the solution conformations of the two proteins are very similar. Negligible Δδ values are observed for all residues of the cap domain and small Δδ values are noted for the majority of residues present in the core domain (Fig. 4). However, some larger Δδ values (0.08 < Δδ < 1.65 ppm) are observed primarily for two contiguous residue segments (D133–A153 and G174–S181), which are located within ~ 8 Å of the P146A mutation site. For a conservative single site amino acid substitution in a protein, chemical shift perturbations caused by changes in the local chemical environment are usually restricted to the immediate vicinity (within ~ 4 Å) of the mutation site (Baxter et al. 2017). Here, the size and the more widespread distribution of the chemical shift changes, together with a propagation of effects through several secondary structure elements, strongly suggest that the conformation of the D137–A147 loop is different in the two proteins. Conformational heterogeneity in this loop is observed when comparing crystal structures of substrate-free βPGMWT (e.g. PDB: 1ZOL versus 2WHE; Zhang et al. 2005; Baxter et al. 2010). The source of this difference in structure is most probably associated with the isomerisation state of the K145–X146 peptide bond. For βPGMWT, all the reported crystal structures have a cis K145–P146 peptide bond and this isomer is also populated in solution as P146 δ13Cβ = 34.8 ppm (Shen and Bax 2010). For βPGMP146A, a regular trans K145–A146 peptide bond is adopted according to TALOS-N, as expected on the basis of the conformational properties of alanine.

A chemical shift comparison between substrate-free βPGMP146A and substrate-free βPGMWT. a Histogram of residue-specific chemical shift changes calculated between βPGMP146A and βPGMWT (BMRB: 7235; Baxter et al. 2006) as Δδ|HN + 0.12*N| = [Δδ2HN + (0.12 × ΔδN)2]1/2, where ΔδX = δX:βPGM:P146A − δX:βPGM:WT and X = 1HN or 15N nuclei of the backbone amide group. Secondary structure elements from substrate-free βPGMWT (PDB: 2WHE) are indicated as bars for α-helices and arrows for β-strands at the top of the panel. Proline residues, the location of the P146A mutation site and unassigned residues in the 1H–15N TROSY spectra of βPGMP146A and βPGMWT are shown as green, red, purple and cyan rectangles, respectively at the top of the panel. b Structure of substrate-free βPGMWT (PDB: 2WHE) with residues coloured according to Δδ|HN + 0.12*N| (Δδ|HN + 0.12*N| > 0.08 ppm) with the intensity of colour and thickness of the backbone corresponding to larger values. The location of the P146A mutation site is highlighted with a red backbone and the catalytic magnesium ion is shown as a green sphere
In conclusion, the isomerisation state of the K145–X146 peptide bond appears to lead to a difference in the active site dynamics, where the cis conformation triggers millisecond exchange for residues of the hinge (V12–T16), the nucleophile (D8) and residues responsible for coordinating the transferring phosphate group in the proximal site (D8 and S114–S116), and residues of the D137–A147 loop (V141–A142 and K145). These occur in addition to millisecond dynamics for residues involved in the coordination of the catalytic MgII ion and for the L44–L53 loop responsible for substrate discrimination and binding.
References
Allen KN, Dunaway-Mariano D (2004) Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem Sci 29:495–503. https://doi.org/10.1016/j.tibs.2004.07.008
Barrozo A, Liao Q, Esguerra M, Marloie G, Florian J, Williams NH, Kamerlin SCL (2018) Computer simulations of the catalytic mechanism of wild-type and mutant β-phosphoglucomutase. Org Biomol Chem 16:2060–2073. https://doi.org/10.1039/C8OB00312B
Baxter NJ, Olguin LF, Goličnik M, Feng G, Hounslow AM, Bermel W, Blackburn GM, Hollfelder F, Waltho JP, Williams NH (2006) A Trojan horse transition state analogue generated by MgF3− formation in an enzyme active site. Proc Natl Acad Sci USA 103:14732–14737. https://doi.org/10.1073/pnas.0604448103
Baxter NJ, Blackburn GM, Marston JP, Hounslow AM, Cliff MJ, Bermel W, Williams NH, Hollfelder F, Wemmer DE, Waltho JP (2008) Anionic charge is prioritized over geometry in aluminum and magnesium fluoride transition state analogs of phosphoryl transfer enzymes. J Am Chem Soc 130:3952–3958. https://doi.org/10.1021/ja078000n
Baxter NJ, Hounslow AM, Bowler MW, Williams NH, Blackburn GM, Waltho JP (2009) MgF3− and α-galactose 1-phosphate in the active site of β-phosphoglucomutase form a transition state analogue of phosphoryl transfer. J Am Chem Soc 131:16334–16335. https://doi.org/10.1021/ja905972m
Baxter NJ, Bowler MW, Alizadeh T, Cliff MJ, Hounslow AM, Wu B, Berkowitz DB, Williams NH, Blackburn GM, Waltho JP (2010) Atomic details of near-transition state conformers for enzyme phosphoryl transfer revealed by MgF3− rather than by phosphoranes. Proc Natl Acad Sci USA 107:4555–4560. https://doi.org/10.1073/pnas.0910333106
Baxter NJ, Zacharchenko T, Barsukov IL, Williamson MP (2017) Pressure-dependent chemical shifts in the R3 domain of talin show that it is thermodynamically poised for binding to either vinculin or RIAM. Structure 25:1–11. https://doi.org/10.1016/j.str.2017.10.008
Dai J, Wang L, Allen KN, Rådström P, Dunaway-Mariano D (2006) Conformational cycling in β-phosphoglucomutase catalysis: reorientation of the β-d-glucose 1,6-(bis)phosphate intermediate. Biochemistry 45:7818–7824. https://doi.org/10.1021/bi060136v
Dai J, Finci L, Zhang C, Lahiri S, Zhang G, Peisach E, Allen KN, Dunaway-Mariano D (2009) Analysis of the structural determinants underlying discrimination between substrate and solvent in β-phosphoglucomutase catalysis. Biochemistry 48:1984–1995. https://doi.org/10.1021/bi801653r
Elsässer B, Dohmeier-Fischer S, Fels G (2012) Theoretical investigation of the enzymatic phosphoryl transfer of β-phosphoglucomutase: revisiting both steps of the catalytic cycle. J Mol Model 18:3169–3179. https://doi.org/10.1007/s00894-011-1344-5
Gardner KH, Kay LE (1998) The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu Rev Biophys and Biomol Struct 27:357–406. https://doi.org/10.1146/annurev.biophys.27.1.357
Goličnik M, Olguin LF, Feng G, Baxter NJ, Waltho JP, Williams NH, Hollfelder F (2009) Kinetic analysis of β-phosphoglucomutase and its inhibition by magnesium fluoride. J Am Chem Soc 131:1575–1588. https://doi.org/10.1021/ja806421f
Griffin JL, Bowler MW, Baxter NJ, Leigh KN, Dannatt HRW, Hounslow AM, Blackburn GM, Webster CE, Cliff MJ, Waltho JP (2012) Near attack conformers dominate β-phosphoglucomutase complexes where geometry and charge distribution reflect those of substrate. Proc Natl Acad Sci USA 109:6910–6915. https://doi.org/10.1073/pnas.1116855109
Jin Y, Bhattasali D, Pellegrini E, Forget SM, Baxter NJ, Cliff MJ, Bowler MW, Jakeman DL, Blackburn GM, Waltho JP (2014) α-Fluorophosphonates reveal how a phosphomutase conserves transition state conformation over hexose recognition in its two-step reaction. Proc Natl Acad Sci USA 111:12384–12389. https://doi.org/10.1073/pnas.1402850111
Johnson LA, Robertson AJ, Baxter NJ, Trevitt CR, Bisson C, Jin Y, Wood HP, Hounslow AM, Cliff MJ, Blackburn GM, Bowler MW, Waltho JP (2018) van der Waals contact between nucleophile and transferring phosphorus is insufficient to achieve enzyme transition-state architecture. ACS Catal 8:8140–8153. https://doi.org/10.1021/acscatal.8b01612
Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN (2002a) Caught in the act: the structure of phosphorylated β-phosphoglucomutase from Lactococcus lactis. Biochemistry 41:8351–8359. https://doi.org/10.1021/bi0202373
Lahiri SD, Zhang G, Rådström P, Dunaway-Mariano D, Allen KN (2002b) Crystallization and preliminary X-ray diffraction studies of β-phosphoglucomutase from Lactococcus lactis. Acta Cryst D58:324–326. https://doi.org/10.1107/S0907444901019989
Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN (2003) The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 299:2067–2071. https://doi.org/10.1126/science.1082710
Lahiri SD, Zhang G, Dai J, Dunaway-Mariano D, Allen KN (2004) Analysis of the substrate specificity loop of the HAD superfamily cap domain. Biochemistry 43:2812–2820. https://doi.org/10.1021/bi0356810
Levander F, Andersson U, Rådström P (2001) Physiological role of β-phosphoglucomutase in Lactococcus lactis. Appl Environ Microbiol 67:4546–4553. https://doi.org/10.1128/AEM.67.10.4546-4553.2001
Marcos E, Field MJ, Crehuet R (2010) Pentacoordinated phosphorus revisited by high-level QM/MM calculations. Proteins 78:2405–2411. https://doi.org/10.1002/prot.22758
Qian N, Stanley GA, Hahn-Hägerdal B, Rådström P (1994) Purification and characterization of two phosphoglucomutases from Lactococcus lactis subsp. lactis and their regulation in maltose- and glucose-utilizing cells. J Bacteriol 176:5304–5311. https://doi.org/10.1128/jb.176.17.5304-5311.1994
Qian N, Stanley GA, Bunte A, Rådström P (1997) Product formation and phosphoglucomutase activities in Lactococcus lactis: cloning and characterization of a novel phosphoglucomutase gene. Microbiology 143:855–865. https://doi.org/10.1099/00221287-143-3-855
Reed MAC, Hounslow AM, Sze KH, Barsukov IG, Hosszu LLP, Clarke AR, Craven CJ, Waltho JP (2003) Effects of domain dissection on the folding and stability of the 43 kDa protein PGK probed by NMR. J Mol Biol 330:1189–1201. https://doi.org/10.1016/S0022-2836(03)00625-9
Shen Y, Bax A (2010) Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J Biomol NMR 46:199–204. https://doi.org/10.1007/s10858-009-9395-y
Shen Y, Bax A (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 56:227–241. https://doi.org/10.1007/s10858-013-9741-y
Tremblay LE, Zhang G, Dai J, Dunaway-Mariano D, Allen KN (2005) Chemical confirmation of a pentavalent phosphorene in complex with β-phosphoglucomutase. J Am Chem Soc 127:5298–5299. https://doi.org/10.1021/ja0509073
Webster CE (2004) High-energy intermediate or stable transition state analogue: theoretical perspective of the active site and mechanism of β-phosphoglucomutase. J Am Chem Soc 126:6840–6841. https://doi.org/10.1021/ja049232e
Zhang G, Dai J, Wang L, Dunaway-Mariano D, Tremblay LW, Allen KN (2005) Catalytic cycling in β-phosphoglucomutase: a kinetic and structural analysis. Biochemistry 44:9404–9416. https://doi.org/10.1021/bi050558p
Acknowledgements
This research was supported by the Consejo Nacional de Ciencia y Tecnologia, Mexico (CONACYT; F.A.C.N.—Grant No. 472448) and the Biotechnology and Biological Sciences Research Council (BBSRC; N.J.B.—Grant No. BB/M021637/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cruz-Navarrete, F.A., Baxter, N.J., Wood, H.P. et al. 1H, 15N and 13C backbone resonance assignments of the P146A variant of β-phosphoglucomutase from Lactococcus lactis in its substrate-free form. Biomol NMR Assign 13, 349–356 (2019). https://doi.org/10.1007/s12104-019-09904-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-019-09904-y