
Abstract
Portal hypertension develops as a result of increased intrahepatic vascular resistance often caused by chronic liver disease that leads to structural distortion by fibrosis, microvascular thrombosis, dysfunction of liver sinusoidal endothelial cells (LSECs), and hepatic stellate cell (HSC) activation. While the basic mechanisms of LSEC and HSC dysregulation have been extensively studied, the role of microvascular thrombosis and platelet function in the pathogenesis of portal hypertension remains to be clearly characterized. As a secondary event, portal hypertension results in splanchnic and systemic arterial vasodilation, leading to the development of a hyperdynamic circulatory syndrome and subsequently to clinically devastating complications including gastroesophageal varices and variceal hemorrhage, hepatic encephalopathy from the formation of portosystemic shunts, ascites, and renal failure due to the hepatorenal syndrome. This review article discusses: (1) mechanisms of sinusoidal portal hypertension, focusing on HSC and LSEC biology, pathological angiogenesis, and the role of microvascular thrombosis and platelets, (2) the mesenteric vasculature in portal hypertension, and (3) future directions for vascular biology research in portal hypertension.
Similar content being viewed by others


Introduction
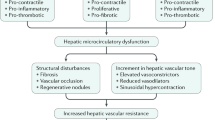
As a disorder of portal venous pressure, portal hypertension can be conceptualized using the hydraulic derivation of Ohm’s Law (pressure = flow × resistance) [1], composed of variables grounded in basic vascular biology. Initially, portal hypertension develops as a result of increased intrahepatic vascular resistance most commonly [2] caused by chronic liver disease leading to multiple pathological events in the sinusoidal circulation, such as structural distortion by fibrosis, microvascular thrombosis, dysfunction of liver sinusoidal endothelial cells (LSECs), and hepatic stellate cell (HSC) activation [3,4,5]. While the basic mechanisms of LSEC and HSC dysregulation have been extensively explored, clarification of the role of microvascular thrombosis and platelet function in the pathogenesis of portal hypertension lags behind.
As a secondary event, portal hypertension leads to splanchnic and systemic arterial vasodilation, contributing to increased splanchnic blood flow to the liver and increased portal pressure despite collateral formation [3,4,5,6,7]. An excessive vasodilation of the mesenteric arteries facilitates this hyperdynamic circulation, and along with increasing blood flow to portosystemic collaterals results in clinically devastating complications including gastroesophageal varices and variceal hemorrhage, hepatic encephalopathy from the formation of portosystemic shunts, ascites, and renal failure due to the hepatorenal syndrome [8,9,10].
In this review article, we discuss the following topics: (1) mechanisms of sinusoidal portal hypertension, focusing on HSC and LSEC biology, pathological angiogenesis, and the role of microvascular thrombosis and platelets, (2) the mesenteric vasculature in portal hypertension, and (3) future directions for vascular biology research in portal hypertension.
Mechanisms of sinusoidal portal hypertension
Hepatic sinusoids are small blood vessels that comprise of the liver microcirculation. Blockage of sinusoids and the resulting increased hepatic vascular resistance to portal venous flow is the primary cause of portal hypertension. Sinusoids consist of LSECs and are circumscribed by HSCs, which are essential for sinusoidal function and key in the development of portal hypertension. Thrombosis is another important factor that disturbs the hepatic microcirculation and increases hepatic vascular resistance. In this section, we discuss the mechanisms of sinusoidal portal hypertension with a focus on HSCs, LSECs, and thrombosis.
Hepatic stellate cell biology
Fibrosis and architectural distortion
Hepatic fibrosis is the first factor to be considered in explaining the increased resistance in the cirrhotic liver. In a classic paper, Bhathal and Grossman demonstrated through vasodilator challenges in an animal model of cirrhosis that 80 % of the increased resistance to portal flow is due to architectural distortion in the cirrhotic liver, while 20 % is due to a reversible, hypercontractile phenotype [11]. While a complete examination of the pathobiology of hepatic fibrosis is outside the scope of this review, this is an area of liver disease with constant and exciting new advances. The key pathway of fibrosis in the liver is proinflammatory signaling causing activation of HSCs and thereby leading to extracellular matrix deposition. As recently reviewed by experts in the field, the understanding of this pathway has led in turn to an understanding of its reversal and the regression of fibrosis, which holds great therapeutic potential for chronic liver disease and portal hypertension [12, 13].
Hepatic stellate cells as pericytes
Hepatic stellate cells (HSCs) are positioned in the space of Disse. They are thus thought to play another role in portal hypertension beyond fibrosis as a possible hepatic pericyte, a perivascular nonendothelial cell with myriad functions including regulation of flow through smooth muscle-like contractility, formation of extracellular matrix (as above), and regulation of endothelial proliferation [14,15,16,17]. This observation arises from work demonstrating that HSCs “activate” and acquire a myofibroblast-like phenotype with acquisition of alpha-smooth muscle actin during the response to liver injury [18], as well as exhibit a contractile phenotype, shown in a gel contraction assay using activated HSCs from a rat model of toxic liver injury [19]. Thus HSCs, through perivascular contraction, are thought to be key contributors to the dynamic and reversible component of portal hypertension in cirrhosis.
Regulation of HSC contraction
Endothelin signaling
A key pathway regulating the HSC contractile phenotype is endothelin signaling. Endothelin binds to G-protein coupled receptors endothelin A (ETA) and endothelin B (ETB), which are typically found on vascular smooth muscle cells and endothelial cells, respectively. Endothelin-1 (ET-1) is the major subtype relevant to liver disease and is preferentially bound to ETA more so than the other two subtypes ET-2 and ET-3 [20]. Levels of endothelin-1 protein are elevated in the setting of liver injury along with ET-1 messenger RNA (mRNA), demonstrating increased production in this setting [21]. Whereas in the normal liver the endothelial cells produce the majority of ET-1, liver injury shifts this production primarily to HSCs [22], which also markedly upregulate ETA and ETB receptors [23, 24], suggesting increased sensitivity to this signal. ET-1 has been shown to induce contraction of the sinusoidal vasculature [25], and antagonism of ETA has been shown to reduce portal pressure in a cirrhotic animal model [26]. Studying the mechanism of ET-1-mediated HSC contraction has yielded the insight that it functions through both a pathway of increasing intracellular calcium, leading to myosin light chain kinase activation and myosin light chain phosphorylation, and via Rho-kinase and protein kinase C pathways leading to inhibition of myosin light chain dephosphorylation by myosin light chain phosphatase and enhanced calcium sensitivity [27].
Other inducers of HSC contraction
Other mechanisms of HSC contraction have been elucidated as well, including C-X-C chemokine receptor 4 (CXCR4) expressed on activated stellate cells. Chemokine (C-X-C motif) ligand 12 (CXCL12) binds CXCR4 and leads to increased myosin light chain phosphorylation and contractility in a Rho kinase-dependent manner, which can be reversed by a Rho kinase inhibitor [28]. Another interesting recent finding is that ammonia may play a direct role in HSC contractility. HSCs exposed to ammonia demonstrated a more contractile phenotype than controls on gel contraction assay and produced higher levels of myosin IIa, which is important for contraction, in a dose-dependent manner [29]. Preclinical data are also emerging on the role of relaxin and its receptor in reducing HSC contraction and portal pressure [30]. A recent study has also pointed to a role for the farnesoid X receptor (FXR) in regulation of the endothelin pathway and HSC contractility, demonstrating that FXR stimulation decreases ET-1 levels and phosphorylated moesin, a biochemical marker of HSC contractility, as well as portal pressure in an animal model of cirrhosis with portal hypertension, though the exact mechanism is uncertain [31].
Liver sinusoidal endothelial cell biology
Fenestration and capillarization
Liver sinusoidal endothelial cells (LSECs) are distinct from endothelial cells elsewhere in the liver, as well as elsewhere in the body. Their most distinguishing feature is fenestration, with fenestrae measuring approximately 0.1 microns organized into groups of sieve plates and thought to facilitate the transport of macromolecules from the hepatic sinusoids to the space of Disse, where they can interact with hepatocytes. Another characteristic setting LSECs apart from endothelial cells in other organs is the lack of a basement membrane, which again maximizes permeability between the lumen of the sinusoid and the space of Disse [32].
LSECs lose their fenestrae and develop a basement membrane as a consequence of liver fibrosis, and become “capillarized” [33, 34]. Vascular endothelial growth factor (VEGF) has been shown to be a key factor in maintaining the endothelial fenestrae, and increasing concentrations of the molecule can increase the porosity of LSECs [35]. The VEGF signaling necessary for maintaining LSEC phenotype, or maintenance of their fenestrae without a basement membrane, is thought to come from HSCs and hepatocytes, since coculture of HSCs or hepatocytes restores this phenotype that is lost with anti-VEGF antibody. This VEGF maintenance of normal LSEC phenotype is dependent on nitric oxide production and is blocked by the NO synthase inhibitor N ω-nitro-l-arginine methyl ester hydrochloride (L-NAME) [36]. Removal of VEGF signaling in an animal model, via a transgenic system in which liver-specific secretion of a soluble VEGF decoy receptor sequesters endogenous VEGF, has been shown to lead to loss of LSEC fenestration and result in portal hypertension and HSC activation independent of hepatic parenchymal damage, with reversal of portal hypertension with restoration of VEGF [37]. In addition, the composition of collagen in the space of Disse may play a role in maintenance or loss of endothelial cell fenestration [38]. Further, a role of lipid rafts in regulation of fenestration has also been elucidated. Utilizing superresolution fluorescence microscopy, researchers recently demonstrated an inverse relationship between areas of the endothelial cell with lipid rafts and areas of the membrane with fenestration. Additionally, it was shown that inhibiting lipid raft formation via 7-ketocholesterol or actin disruption increased fenestration, and increasing raft formation with a low concentration of Triton X-100 decreased fenestration [39].
Crosstalk between LSECs and HSCs
The communication between LSECs and HSCs is important in the pathogenesis of portal hypertension, as evidenced by the fact that loss of LSEC phenotype can be permissive for HSC activation [40]. Research into LSEC and HSC communication demonstrated that an isoform of fibronectin produced by LSECs in a bile duct ligation model of liver damage was able to activate HSCs [41], though a subsequent study revealed it to be a factor important for HSC motility but not differentiation to a myofibroblast phenotype [42]. Recent literature has also revealed a mechanism for LSECs to communicate with HSCs through signaling via exosomes containing sphingosine kinase-1 (SK1) and its product sphingosine-1 phosphate, providing a signal for HSC migration [43], which is closely tied to their activated phenotype. Conversely, LSEC signaling can also be responsible for the maintenance of HSC quiescence. LSEC induction of HSC deactivation has been shown to be possible via paracrine signaling via the Kruppel-like factor 2 (KLF2)–nitric oxide–guanylate cyclase pathway in endothelial cells [44]. Various pathways of LSEC/HSC crosstalk are depicted in Fig. 1.

LSEC/HSC crosstalk. Liver injury leads LSECs to produce the EIIIA isoform of fibronectin, which signals to HSCs through integrin α9β1 to promote motility, which is important for their activated phenotype [41, 42]. LSECs also signal to promote HSC motility through sphingosine kinase 1–sphingosine-1-phosphate (SK1–S1P)-containing exosomes, which adhere to HSCs via fibronectin binding to an integrin receptor [43]. Nitric oxide (NO) production by sinusoidal endothelial cells is important in maintaining HSC quiescence, with a Kruppel-like factor (KLF) 2 pathway enhancing NO and guanylate cyclase production [44], and NO production from LSECs also inhibiting the Rac/Rho pathway in HSCs [47, 48]. NO also causes HSC apoptosis, and may thereby limit the number of activated HSCs in the liver [49]. Vit A, vitamin A
Nitric oxide, which is produced by LSECs and plays a direct role in regulation of vascular tone in the liver as discussed below, also plays a key role in LSEC/HSC crosstalk. LSECs are able to induce HSC reversion from activation to quiescence via an NO-dependent mechanism [45], which may have relevance in diseases such as alcoholic liver injury and nonalcoholic fatty liver disease, in which loss of normal LSEC phenotype has been shown to occur before fibrosis [40]. Studies relevant to this NO-dependent mechanism have shown that NO donors are able to inhibit proliferation and chemotaxis of activated HSCs in response to platelet-derived growth factor by disruption of its intracellular signaling pathway via prostaglandin E2-mediated effects [46], and that NO inhibits HSC migration via cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PKG)-mediated inhibition of the Rac1 pathway [47, 48]. Nitric oxide signaling has also been shown to induce HSC apoptosis [49] via a caspase-independent mechanism possibly related to increased mitochondrial oxidative stress and increased mitochondrial membrane permeability, along with a possible lysosomal stress component.
Endothelial nitric oxide: function and dysfunction
Nitric oxide (NO) is a crucially important regulator of normal hepatic vascular tone and portal pressure [50], and the source of NO in the hepatic vasculature is the LSECs and endothelial cells of blood vessels. These cells express an endothelial nitric oxide synthase (eNOS) and produce a baseline level of NO. Production of NO increases in response to flow [51] and can also be upregulated by VEGF [52]. Studies have demonstrated that, although LSECs in the cirrhotic liver contain similar amounts of eNOS as in the normal liver, it is dysfunctional under these pathologic conditions and there is diminished NO release in disease [53]. Additionally, LSECs in cirrhosis have a diminished ability to respond to increases in flow with increased NO production compared with the healthy state [54]. This derangement leads to impaired vasodilation in the hepatic microcirculation in cirrhosis and is an important contributor to sinusoidal portal hypertension. The regulation of eNOS function involves multiple players acting in concert, with stimulatory signaling found to occur via phosphorylation by the protein kinase Akt [55], and subsequent work elucidating the G-protein coupled-receptor kinase interactor-1 (GIT1) as a facilitator of Akt-dependent eNOS activation [56, 57]. The function of eNOS may also be inhibited by binding to caveolin-1 in an interaction that can in turn be disrupted by calmodulin [58].
Because endothelial dysfunction leads to increased resistance in the sinusoidal microcirculation and promotes activation of HSCs, a pharmacological approach that reverses the dysfunctional LSEC phenotype could be an effective therapeutic strategy. An emerging example is the use of statins. Studies have shown that statins ameliorate portal hypertension in cirrhotic patients [59] and experimental models of portal hypertension [60, 61]. These studies demonstrated that statins improve endothelial dysfunction by increasing NO bioavailability in the sinusoidal microcirculation. Several candidate mechanisms for this observed effect have been proposed. One is the ability of statins to inhibit synthesis of isoprenoids, which are necessary for membrane anchoring and activation of small guanine triphosphatases (GTPases), such as RhoA. Given that RhoA/Rho-kinase signaling could downregulate eNOS activity [62] and expression [63], statins, by decreasing RhoA activity, could enhance NO bioavailability and decrease intrahepatic vascular resistance [61]. Another potential mechanism is that statins increase activity of Akt/protein kinase B, which phosphorylates and activates eNOS, thereby increasing NO bioavailability [60]. In addition to reducing endothelial cell dysfunction, statins could target the RhoA/Rho-kinase pathway in pericytes (e.g., activated HSCs) and decrease their contractile phenotype, thereby lowering intrahepatic resistance and reducing portal hypertension [61].
Angiogenesis
Angiogenesis, or the process of new blood vessel formation from preexisting vascular beds, has been implicated in portal hypertension as well. Hepatic angiogenesis could cause irregular intrahepatic circulatory routes and thus could increase intrahepatic resistance. Pathological angiogenesis in the splanchnic circulation has been thought to worsen portal hypertension because increased vasculature created via angiogenesis could enhance blood flow to the portal venous system, thereby increasing portal pressure. Blocking pathological angiogenesis by kinase inhibitors, such as sorafenib [64, 65], sunitinib [66], imatinib [67], pioglitazone [68], and the combination of Gleevec and rapamycin [69], has been shown to ameliorate portal hypertension in experimental models.
In addition, the Notch1 signaling pathway, which is known to be important in endothelial cell differentiation and vascular development, has been shown to be important in portal hypertension, with knockout of the pathway in the liver leading to nodular regenerative hyperplasia (NRH), a common cause of noncirrhotic portal hypertension. Interestingly, knockout animals developed portal hypertension even before the onset of NRH, which was thought to be caused by a disordered intrahepatic vasculature [70]. Angiogenesis has also been shown to be associated with fibrosis progression in the liver [71, 72], although this relationship is complex and could be causative or correlative due to hypoxia, with modulation of angiogenesis not having a predictable effect on liver fibrosis [73].
Microvascular thrombosis/platelets
Role of thrombosis in the pathogenesis of portal hypertension
The study of intrahepatic portal hypertension is evolving to include the study of platelets and thrombosis as important contributors to its pathophysiology. Ian Wanless and others were instrumental contributors in this area, observing what they termed “parenchymal extinction” accounting for fibrosis progression due to intrahepatic vascular thrombosis [74]. Though cirrhosis had been previously felt to confer a bleeding tendency, more advanced and physiologic tests to assess coagulation status [75] and systematic studies of bleeding complications [76] have led to a growing consensus that hemostatic status in cirrhosis is delicately rebalanced or perhaps even prothrombotic. These observations set the stage for exploration of the role of thrombosis in sinusoidal portal hypertension.
Animal studies
Animal studies lend support to the importance of thrombosis in fibrosis progression and portal hypertension. An early study utilized mice on a susceptible genetic background infected with the murine hepatitis virus, an RNA coronavirus that can cause a spectrum of liver disease including fulminant hepatic failure. These mice exhibited thrombi in the hepatic microvasculature concordant with viral liver injury [77], suggesting a link between sinusoidal thrombosis/microvascular blockage and liver damage that was supported in further studies [78]. Other animal studies using a model of carbon tetrachloride (CCl4)-induced liver damage demonstrated sinusoidal deposition of fibrin/fibrinogen and fibronectin in the damaged liver in the short-term, and deposition in fibrous septa in long-term liver damage, leading to the hypothesis that clotting was an important step in the fibrotic response of the liver [79]. In more mechanistic preclinical studies, administering a novel thrombin antagonist (SR182289) in a CCl4 model was shown to decrease liver fibrosis [80], and mice deficient in the prothrombinase fgl2/fibroleukin, responsible for cleaving prothrombin to thrombin and ultimately the deposition of fibrin by this pathway, had reduced fibrin deposition and necrosis in a model of viral hepatitis [81].
Low-molecular-weight heparins as anticoagulants were studied and shown to reduce fibrosis in a bile duct ligation model, a thioacetamide model (in which aspirin was also shown to reduce fibrosis), and a CCl4 model of hepatic fibrosis [82,83,84]. Again in a CCl4 model, factor V Leiden homozygous mice had more hepatic fibrosis than wild-type mice, and anticoagulation with warfarin reduced hepatic fibrosis in wild-type mice compared with control [85]. Notably in this study, warfarin treatment did not have an antifibrotic effect in factor V Leiden homozygous mice, which the authors speculate was because the dose was not adequate to overcome the profibrotic phenotype. In a recent study mechanistically linking portal hypertension with hepatic sinusoidal thrombosis and fibrosis, an inferior vena cava (IVC) ligation model of post-hepatic portal hypertension resulted in sinusoidal thrombosis and hepatic fibrosis, which was ameliorated by pharmacologic treatment with warfarin and genetic inhibition of the clotting cascade, and fibrin was shown to promote extracellular matrix deposition by HSCs [86]. An important recent paper in rat models of liver injury/fibrosis with CCl4 or thioacetamide with and without enoxaparin demonstrated reduced portal pressure, reduced HSC activation, reduced fibrosis, and reduced fibrin deposition in treated animals as compared with control animals, pointing to a role for anticoagulation in reducing classic structural and dynamic mechanisms of portal hypertension, possibly via reduction of thrombosis [87]. Rivaroxaban has also been shown in thioacetamide and CCl4 models of cirrhosis in the rat to reduce portal pressure, likely through a combination of reductions in HSC activation, endothelial dysfunction, and microvascular thrombosis rather than fibrosis [88].
Platelets
Platelets are intimately associated with the typical physiology of vascular thrombosis, and investigation into their biology in cirrhosis is an emerging area of careful study. Initially, the assessment of the cirrhotic platelet was that it was dysfunctional and predisposed the patient to a bleeding tendency [89, 90]. More recently, however, investigators have found that the activity of cirrhotic platelets in hemostasis and thrombosis is potentially preserved [91] or even increased [92], although there are conflicting data [93]. As recently reviewed [94], the function of platelets in various types of liver injury is quite complex, with multiple situation-specific factors determining whether platelets will play a profibrotic role versus an antifibrotic, pro-regenerative role. Of particular relevance to portal hypertension, platelet-derived serotonin in a model of viral hepatitis results in hepatic microcirculatory dysfunction in the sinusoids leading to reduced flow [95], consistent with other studies demonstrating serotonin-mediated low sinusoidal flow reversed by serotonin receptor antagonism [96, 97]. This effect may be mediated by serotonin-induced calcium influx into sinusoidal endothelial cells and myosin light chain phosphorylation causing fenestral contraction [98, 99], or by serotonin-mediated HSC activation, possibly through increasing intracellular calcium [100, 101].
Given that nonalcoholic steatohepatitis (NASH) is associated with the metabolic syndrome, with its associated platelet dysfunction resulting in a hypercoagulable platelet with increased expression of glycoprotein (GP) IIb/IIIa receptors and resistance to antiaggregating stimuli such as NO and prostaglandins [102], platelet function in this disease is of particular interest. In an animal model of NASH, antiplatelet drugs such as aspirin, ticlopidine (a thienopyridine adenosine diphosphate receptor antagonist) [103], and cilostazol (a phosphodiesterase III inhibitor) were all found to reduce hepatic steatosis, inflammation, and fibrosis, and cilostazol also led to increased eNOS production measured by mRNA expression. However, cilostazol had the largest antifibrotic effect and is postulated to impact lipid and glucose metabolism in addition to its antiplatelet activity, so these observations may not be entirely because of antithrombotic effects [104]. The putative roles of platelets and thrombosis in portal hypertension are shown schematically in Fig. 2.

Sinusoidal thrombosis and portal hypertension. Sinusoidal thrombosis and platelet aggregation/activation contribute to portal hypertension through multiple pathways. Hepatocyte damage results in release of tissue factor (TF) [132], which promotes activation of factor VIIa and Xa and ultimately prothrombin conversion to thrombin, which converts fibrinogen to fibrin in an intravascular thrombus [133]. Parenchymal extinction may occur due to the presence of this thrombus. Fgl2/fibroleukin found on endothelial cells may also promote fibrin clot formation [81]. Thrombin may activate HSCs through protease-activated receptor 1 (PAR1) [134] as well as promote platelet aggregation/activation through PAR4 [135], with platelet adhesion to the endothelium mediated by glycoprotein (GP) Ib and the integrins αIIβ3 and αVβ3 [136]. Platelets can then activate HSCs through release of serotonin [100, 101] and platelet-derived growth factor B (PDGF-B) [137]. Serotonin also leads to increased myosin light chain phosphorylation (P-MLC) in LSECs, fenestral contraction, and microvascular dysfunction [98, 99]. Sinusoidal fibrin may also reach HSCs and promote fibronectin deposition into extracellular matrix (ECM) via αVβ1 integrin [86]. Vit A, vitamin A, 5HT2R, 5-HT2 receptor
Future experimentation and clinical trials will continue to expand our knowledge regarding thrombosis, cirrhotic platelet biology, and portal hypertension. In particular, the ongoing Multicenter Prospective Randomized Trial of the Effectiveness of Rivaroxaban (a direct factor Xa inhibitor) [103] on Survival and Development of Complications of Portal Hypertension in Patients with Cirrhosis (CIRROXABAN) will provide evidence for any effect of anticoagulation to alter the course of cirrhosis and portal hypertension (ClinicalTrials.gov NCT02643212). Antithrombotic and antiplatelet drugs that have been shown to have some role in ameliorating portal hypertension, fibrosis, or hepatic decompensation are depicted along with their site(s) of action in the coagulation cascade in Fig. 3.

The coagulation cascade. The coagulation cascade, with the points of action of antithrombotic/antiplatelet compounds that have been demonstrated to have antifibrotic effects, reduce portal hypertension, or have clinical benefits in liver disease. Warfarin, low-molecular-weight heparin (LMWH), cilostazol, ticlopidine, aspirin, and SR182289 have been shown to have antifibrotic effects in experimental models. Rivaroxaban and LMWH have both been directly shown to reduce portal hypertension in experimental models, and LMWH reduced hepatic decompensation in cirrhotic patients in one clinical study
The mesenteric vasculature in portal hypertension
In addition to the liver vasculature, the mesenteric vasculature plays a key role in portal hypertension. Foundational understanding of the physiology of this vascular bed comes from classic studies by Groszmann and others which demonstrated that, even with the development of portosystemic collaterals, the splenchnic circulation was hyperdynamic in portal hypertension [105, 106]. Accordingly, portal pressure remains elevated due to the increased splanchnic flow to the liver [4, 5]. Excessive vasodilation of splanchnic arteries mediated by overproduction of NO by eNOS contributes to this increased flow. A study using rats with partial portal vein ligation showed higher eNOS levels and increased NO production precede the development of the hyperdynamic circulation [107] and that VEGF is an important mediator of eNOS activation [52]. Other studies have demonstrated higher eNOS activity leading to NO overproduction in the mesenteric vasculature in portal hypertension via an Akt-dependent pathway leading to eNOS phosphorylation (an active form of eNOS) [108].
Other mechanisms by which splanchnic vasodilation occurs in cirrhosis are via ACE2 conversion of angiotensin (Ang) II to Ang (1–7) and subsequent activation of the Mas receptor [109]. Other pathways via carbon monoxide, prostacyclin, endocannabinoids, adrenomedullin, and endothelium-derived hyperpolarizing factor play a role in this pathology as well [5]. As previously reviewed [1], arterial thinning [110] and smooth muscle hypocontractility also promote the disordered physiology of the mesenteric vasculature in portal hypertension.
The impaired sympathetic nervous system has also been implicated in hypocontractility of mesenteric arteries and the development of the hyperdynamic circulatory syndrome in portal hypertensive rats [111,112,113]. These animals exhibited mesenteric sympathetic nerve atrophy/regression, but administration of agents that inhibit these nerve disorders (e.g., capsaicin [114] and gambogic amide [115]) ameliorated the hyperdynamic circulatory syndrome. These impaired neuronal functions were also associated with decreased release of neuropeptide Y, a neurotransmitter and potent vasoconstrictor produced by the sympathetic nervous system [116]. Administration of exogenous neuropeptide Y decreased excessive vasodilation of mesenteric arteries of portal hypertensive rats, indicating that hypocontractility of mesenteric arteries is at least in part mediated by an impaired production of neural factors [116, 117].
More recent studies have explored agents for ameliorating the hyperdynamic and hypocontractile mesenteric vascular phenotype in cirrhosis using thalidomide [118] and an arginine vasopressin receptor 1a partial agonist [119].
Future directions
Several important biological systems that ought to be relevant to the pathophysiology of portal hypertension have not yet been adequately explored. Of particular interest are immune cells, platelets, and the lymphatic vascular system.
Immune cells
It is not known whether immune cells are directly related to the development of portal hypertension. However, since immune cells play an important role in hepatic fibrogenesis and fibrosis is a major cause of portal hypertension, they can be considered as a key regulator of it. The role of macrophages is particularly noteworthy. Macrophages have been shown to facilitate hepatic fibrosis and pathological angiogenesis in fibrotic/cirrhotic mice, and conversely inhibition of macrophage infiltration reduced these pathological events [120]. A direct interaction of macrophages with endothelial cells was also shown, suggesting a paracrine signaling relationship between them [121].
In fact, LSECs serve as a layer for adhesion of a variety of resident immune cell populations by expressing various chemokine receptors and adhesion molecules [122]. LSECs also regulate immune cell functions and phenotypes, such as T cell differentiation and macrophage polarization. Portal hypertension is associated with LSEC dysfunction, which could thus affect immune cell function. In addition to regulation of intrahepatic vascular tone, elucidation of how LSEC dysfunction influences immune cell function is important to advance our understanding of hepatic vascular biology.
Platelets
As discussed above, platelet biology in cirrhosis has received increasing attention and is becoming an area of active investigation, with our understanding of platelet function and fibrinolysis remaining insufficient [123]. More studies to define coagulation states in cirrhotic patients are needed using reliable methodologies and with consideration of their diverse clinical presentations.
Platelet adhesion and aggregation are critical steps in coagulation and thrombosis, and key to these functions is the interaction of platelets with endothelial cells [124]. Endothelial cells produce potent inhibitors of platelet activation, such as NO and prostacyclin, along with an adenosine diphosphatase (ADPase) enzyme, CD39, which decomposes ADP, a strong activator of platelets. When endothelial dysfunction or damage occurs, these platelet inhibitory functions are impaired, and instead generation and deposition of platelet adhesion factors, such as von Willebrand factor, collagen, and fibrinogen, occur on the endothelial cell surface, facilitating platelet adhesion and activation. Given that LSEC dysfunction is a well-known condition in liver cirrhosis and portal hypertension [6, 7, 125], an interaction between LSECs and platelets as it relates to thrombosis should be better defined. Platelets also influence LSEC function, modulating their production of growth factors such as interleukin (IL)-6 [126]. Additionally, platelets produce transforming growth factor beta (TGFβ) [124], the most potent factor that causes endothelial-to-mesenchymal transition [127, 128]. This wealth of biology leaves many interesting and important questions as to the role of platelets in the pathogenesis of liver cirrhosis and portal hypertension, especially regarding their interaction with LSECs.
Lymphatic vascular system
The role of lymphatic vessels in the liver is largely unknown. However, an increase in lymphatic vessels has been reported in liver fibrosis, idiopathic portal hypertension, and hepatocellular carcinoma (HCC), suggesting their involvement in these pathological conditions [129,130,131]. There are many questions to be answered including the functional significance of these vessels, their role in promoting or mitigating pathogenesis, mechanisms of lymphangiogenesis, and the relationship between lymphangiogenesis and hemodynamics. Elucidating the mechanisms of lymphangiogenesis associated with portal hypertension will be an interesting area of investigation.
Conclusions
The pathogenesis of portal hypertension is complex, because portal hypertension involves not only the hepatic circulation, but also the hyperdynamic physiology in the systemic and splanchnic circulations. Many pathophysiologic questions remain to be explored, including the roles of immune cells, platelets, and the lymphatic vascular system mentioned above. An understanding of the involvement of such factors will advance our knowledge of portal hypertension in the hope of discovering new therapeutic interventions.
References
Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis—current status and future directions. J Hepatol 2014;61:912–924
Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J Hepatol 2015;62:S121–S130
Iwakiri Y. The molecules: mechanisms of arterial vasodilatation observed in the splanchnic and systemic circulation in portal hypertension. J Clin Gastroenterol 2007;41(Suppl 3):S288–S294
Iwakiri Y. Pathophysiology of portal hypertension. Clin Liver Dis 2014;18:281–291
Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology 2006;43:S121–S131
Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int 2012;32:199–213
Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. J Hepatol 2007;46:927–934
Tetangco EP, Silva R, Lerma E. Portal hypertension: etiology, evaluation, and management. Dis Mon 2016;62(12):411–426
Cavallin M, Fasolato S, Marenco S, Piano S, Tonon M, Angeli P. The treatment of hepatorenal syndrome. Dig Dis 2015;33:548–554
Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2017;65:310–335
Bhathal PS, Grossman HJ. Reduction of the increased portal vascular resistance of the isolated perfused cirrhotic rat liver by vasodilators. J Hepatol 1985;1:325–337
Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64:830–841
Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: concept to treatment. J Hepatol 2015;62:S15–S24
Shepro D, Morel NM. Pericyte physiology. FASEB J 1993;7:1031–1038
Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 2005;7:452–464
Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 2011;118:2906–2917
LaBarbera KE, Hyldahl RD, O’Fallon KS, Clarkson PM, Witkowski S. Pericyte NF-kappaB activation enhances endothelial cell proliferation and proangiogenic cytokine secretion in vitro. Physiol Rep 2015;3(4):e12309
Rockey DC, Boyles JK, Gabbiani G, Friedman SL. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol 1992;24:193–203
Rockey DC, Housset CN, Friedman SL. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J Clin Invest 1993;92:1795–1804
Rockey DC. Vascular mediators in the injured liver. Hepatology 2003;37:4–12
Rockey DC, Fouassier L, Chung JJ, Carayon A, Vallee P, Rey C, et al. Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology 1998;27:472–480
Shao R, Yan W, Rockey DC. Regulation of endothelin-1 synthesis by endothelin-converting enzyme-1 during wound healing. J Biol Chem 1999;274:3228–3234
Rothermund L, Leggewie S, Schwarz A, Thone-Reinecke C, Cho JJ, Bauer C, et al. Regulation of the hepatic endothelin system in advanced biliary fibrosis in rats. Clin Chem Lab Med 2000;38:507–512
Yokomori H, Oda M, Ogi M, Kamegaya Y, Tsukada N, Nakamura M, et al. Enhanced expression of endothelin receptor subtypes in cirrhotic rat liver. Liver 2001;21:114–122
Zhang JX, Pegoli W Jr, Clemens MG. Endothelin-1 induces direct constriction of hepatic sinusoids. Am J Physiol 1994;266:G624–G632
Feng HQ, Weymouth ND, Rockey DC. Endothelin antagonism in portal hypertensive mice: implications for endothelin receptor-specific signaling in liver disease. Am J Physiol Gastrointest Liver Physiol 2009;297:G27–G33
Iizuka M, Murata T, Hori M, Ozaki H. Increased contractility of hepatic stellate cells in cirrhosis is mediated by enhanced Ca2+-dependent and Ca2+-sensitization pathways. Am J Physiol Gastrointest Liver Physiol 2011;300:G1010–G1021
Saiman Y, Agarwal R, Hickman DA, Fausther M, El-Shamy A, Dranoff JA, et al. CXCL12 induces hepatic stellate cell contraction through a calcium-independent pathway. Am J Physiol Gastrointest Liver Physiol 2013;305:G375–G382
Jalan R, De Chiara F, Balasubramaniyan V, Andreola F, Khetan V, Malago M, et al. Ammonia produces pathological changes in human hepatic stellate cells and is a target for therapy of portal hypertension. J Hepatol 2016;64:823–833
Fallowfield JA, Hayden AL, Snowdon VK, Aucott RL, Stutchfield BM, Mole DJ, et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 2014;59:1492–1504
Schwabl P, Hambruch E, Seeland BA, Hayden H, Wagner M, Garnys L, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol 2017;66(4):724–33
Wisse E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J Ultrastruct Res 1970;31:125–150
Bhunchet E, Fujieda K. Capillarization and venularization of hepatic sinusoids in porcine serum-induced rat liver fibrosis: a mechanism to maintain liver blood flow. Hepatology 1993;18:1450–1458
Horn T, Christoffersen P, Henriksen JH. Alcoholic liver injury: defenestration in noncirrhotic livers–a scanning electron microscopic study. Hepatology 1987;7:77–82
Funyu J, Mochida S, Inao M, Matsui A, Fujiwara K. VEGF can act as vascular permeability factor in the hepatic sinusoids through upregulation of porosity of endothelial cells. Biochem Biophys Res Commun 2001;280:481–485
DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol 2004;287:G757–G763
May D, Djonov V, Zamir G, Bala M, Safadi R, Sklair-Levy M, et al. A transgenic model for conditional induction and rescue of portal hypertension reveals a role of VEGF-mediated regulation of sinusoidal fenestrations. PLoS ONE 2011;6:e21478
McGuire RF, Bissell DM, Boyles J, Roll FJ. Role of extracellular matrix in regulating fenestrations of sinusoidal endothelial cells isolated from normal rat liver. Hepatology 1992;15:989–997
Svistounov D, Warren A, McNerney GP, Owen DM, Zencak D, Zykova SN, et al. The relationship between fenestrations, sieve plates and rafts in liver sinusoidal endothelial cells. PLoS ONE 2012;7:e46134
DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015;61:1740–1746
Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol 1994;127:2037–2048
Olsen AL, Sackey BK, Marcinkiewicz C, Boettiger D, Wells RG. Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology 2012;142(928–937):e923
Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, et al. Exosome adherence and internalization by hepatic stellate cells triggers sphingosine 1-phosphate-dependent migration. J Biol Chem 2015;290:30684–30696
Marrone G, Russo L, Rosado E, Hide D, Garcia-Cardena G, Garcia-Pagan JC, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol 2013;58:98–103
Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008;48:920–930
Failli P, De FR, Caligiuri A, Gentilini A, Romanelli RG, Marra F, et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology 2000;119:479–492
Routray C, Liu C, Yaqoob U, Billadeau DD, Bloch KD, Kaibuchi K, et al. Protein kinase G signaling disrupts Rac1-dependent focal adhesion assembly in liver specific pericytes. Am J Physiol Cell Physiol 2011;301:C66–C74
Lee JS, Kang Decker N, Chatterjee S, Yao J, Friedman S, Shah V. Mechanisms of nitric oxide interplay with Rho GTPase family members in modulation of actin membrane dynamics in pericytes and fibroblasts. Am J Pathol 2005;166:1861–1870
Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, et al. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology 2008;47:1983–1993
Mittal MK, Gupta TK, Lee FY, Sieber CC, Groszmann RJ. Nitric oxide modulates hepatic vascular tone in normal rat liver. Am J Physiol 1994;267:G416–G422
Shah V, Haddad FG, Garcia-Cardena G, Frangos JA, Mennone A, Groszmann RJ. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 1997;100:2923–2930
Abraldes JG, Iwakiri Y, Loureiro-Silva M, Haq O, Sessa WC, Groszmann RJ. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state. Am J Physiol Gastrointest Liver Physiol 2006;290:G980–G987
Rockey DC, Chung JJ. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: endothelial dysfunction in portal hypertension. Gastroenterology 1998;114:344–351
Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999;117:1222–1228
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999;399:597–601
Liu S, Premont RT, Rockey DC. G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. J Biol Chem 2012;287:12309–12320
Liu S, Premont RT, Rockey DC. Endothelial nitric-oxide synthase (eNOS) is activated through G-protein-coupled receptor kinase-interacting protein 1 (GIT1) tyrosine phosphorylation and Src protein. J Biol Chem 2014;289:18163–18174
Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem 1997;272:15583–15586
Abraldes JG, Albillos A, Banares R, Turnes J, Gonzalez R, Garcia-Pagan JC, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651–1658
Abraldes JG, Rodriguez-Vilarrupla A, Graupera M, Zafra C, Garcia-Caldero H, Garcia-Pagan JC, et al. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol 2007;46:1040–1046
Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007;46:242–253
Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 2002;22:8467–8477
Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 1998;273:24266–24271
Mejias M, Garcia-Pras E, Tiani C, Miquel R, Bosch J, Fernandez M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology 2009;49:1245–1256
Reiberger T, Angermayr B, Schwabl P, Rohr-Udilova N, Mitterhauser M, Gangl A, et al. Sorafenib attenuates the portal hypertensive syndrome in partial portal vein ligated rats. J Hepatol 2009;51:865–873
Tugues S, Fernandez-Varo G, Munoz-Luque J, Ros J, Arroyo V, Rodes J, et al. Antiangiogenic treatment with sunitinib ameliorates inflammatory infiltrate, fibrosis, and portal pressure in cirrhotic rats. Hepatology 2007;46:1919–1926
Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology 2008;135:671–679
Schwabl P, Payer BA, Grahovac J, Klein S, Horvatits T, Mitterhauser M, et al. Pioglitazone decreases portosystemic shunting by modulating inflammation and angiogenesis in cirrhotic and non-cirrhotic portal hypertensive rats. J Hepatol 2014;60(6):1135–1142
Fernandez M, Mejias M, Garcia-Pras E, Mendez R, Garcia-Pagan JC, Bosch J. Reversal of portal hypertension and hyperdynamic splanchnic circulation by combined vascular endothelial growth factor and platelet-derived growth factor blockade in rats. Hepatology 2007;46:1208–1217
Dill MT, Rothweiler S, Djonov V, Hlushchuk R, Tornillo L, Terracciano L, et al. Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 2012;142(967–977):e962
Corpechot C, Barbu V, Wendum D, Kinnman N, Rey C, Poupon R, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002;35:1010–1021
Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Mockel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014;63:1960–1971
Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol 2010;53:976–980
Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995;21:1238–1247
Tripodi A. Hemostasis abnormalities in cirrhosis. Curr Opin Hematol 2015;22:406–412
De Pietri L, Bianchini M, Montalti R, De Maria N, Di Maira T, Begliomini B, et al. Thrombelastography-guided blood product use before invasive procedures in cirrhosis with severe coagulopathy: a randomized, controlled trial. Hepatology 2016;63:566–573
Levy GA, MacPhee PJ, Fung LS, Fisher MM, Rappaport AM. The effect of mouse hepatitis virus infection on the microcirculation of the liver. Hepatology 1983;3:964–973
MacPhee PJ, Schmidt EE, Keown PA, Groom AC. Microcirculatory changes in livers of mice infected with murine hepatitis virus. Evidence from microcorrosion casts and measurements of red cell velocity. Microvasc Res 1988;36:140–149
Neubauer K, Knittel T, Armbrust T, Ramadori G. Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology 1995;108:1124–1135
Duplantier JG, Dubuisson L, Senant N, Freyburger G, Laurendeau I, Herbert JM, et al. A role for thrombin in liver fibrosis. Gut 2004;53:1682–1687
Marsden PA, Ning Q, Fung LS, Luo X, Chen Y, Mendicino M, et al. The Fgl2/fibroleukin prothrombinase contributes to immunologically mediated thrombosis in experimental and human viral hepatitis. J Clin Invest 2003;112:58–66
Abdel-Salam OM, Baiuomy AR, Ameen A, Hassan NS. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharmacol Res 2005;51:59–67
Assy N, Hussein O, Khalil A, Luder A, Szvalb S, Paizi M, et al. The beneficial effect of aspirin and enoxaparin on fibrosis progression and regenerative activity in a rat model of cirrhosis. Dig Dis Sci 2007;52:1187–1193
Abe W, Ikejima K, Lang T, Okumura K, Enomoto N, Kitamura T, et al. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J Hepatol 2007;46:286–294
Anstee QM, Goldin RD, Wright M, Martinelli A, Cox R, Thursz MR. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemost 2008;6:1336–1343
Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology 2015;61:648–659
Cerini F, Vilaseca M, Lafoz E, Garcia-Irigoyen O, Garcia-Caldero H, Tripathi DM, et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol 2016;64:834–842
Vilaseca M, Garcia-Caldero H, Lafoz E, Garcia-Irigoyen O, Avila M, Reverter JC, et al. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017;65(6):2031–2044
Ordinas A, Escolar G, Cirera I, Vinas M, Cobo F, Bosch J, et al. Existence of a platelet-adhesion defect in patients with cirrhosis independent of hematocrit: studies under flow conditions. Hepatology 1996;24:1137–1142
Rubin MH, Weston MJ, Langley PG, White Y, Williams R. Platelet function in chronic liver disease: relationship to disease severity. Dig Dis Sci 1979;24:197–202
Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006;44:53–61
Raparelli V, Basili S, Carnevale R, Napoleone L, Del Ben M, Nocella C, et al. Low-grade endotoxemia and platelet activation in cirrhosis. Hepatology 2017;65(2):571–81
Potze W, Siddiqui MS, Boyett SL, Adelmeijer J, Daita K, Sanyal AJ, et al. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J Hepatol 2016;65:980–987
Chauhan A, Adams DH, Watson SP, Lalor PF. Platelets: no longer bystanders in liver disease. Hepatology 2016;64:1774–1784
Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med 2008;14:756–761
Ruddell RG, Mann DA, Ramm GA. The function of serotonin within the liver. J Hepatol 2008;48:666–675
Cummings JL, Cilento EV, Reilly FD. Hepatic microvascular regulatory mechanisms. XII. Effects of 5-HT2-receptor blockade on serotonin-induced intralobular hypoperfusion. Int J Microcirc Clin Exp 1993;13:99–112
Brauneis U, Gatmaitan Z, Arias IM. Serotonin stimulates a Ca2+ permeant nonspecific cation channel in hepatic endothelial cells. Biochem Biophys Res Commun 1992;186:1560–1556
Gatmaitan Z, Varticovski L, Ling L, Mikkelsen R, Steffan AM, Arias IM. Studies on fenestral contraction in rat liver endothelial cells in culture. Am J Pathol 1996;148:2027–2041
Park KS, Sin PJ, Lee DH, Cha SK, Kim MJ, Kim NH, et al. Switching-on of serotonergic calcium signaling in activated hepatic stellate cells. World J Gastroenterol 2011;17:164–173
Kim DC, Jun DW, Kwon YI, Lee KN, Lee HL, Lee OY, et al. 5-HT2A receptor antagonists inhibit hepatic stellate cell activation and facilitate apoptosis. Liver Int 2013;33:535–543
Morange PE, Alessi MC. Thrombosis in central obesity and metabolic syndrome: mechanisms and epidemiology. Thromb Haemost 2013;110:669–680
Committee ASoP, Acosta RD, Abraham NS, Chandrasekhara V, Chathadi KV, Early DS, et al. The management of antithrombotic agents for patients undergoing GI endoscopy. Gastrointest Endosc 2016;83:3–16
Fujita K, Nozaki Y, Wada K, Yoneda M, Endo H, Takahashi H, et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008;57:1583–1591
Groszmann R, Kotelanski B, Cohn JN, Khatri IM. Quantitation of portasystemic shunting from the splenic and mesenteric beds in alcoholic liver disease. Am J Med 1972;53:715–722
Kotelanski B, Groszmann R, Cohn JN. Circulation times in the splanchnic and hepatic beds in alcoholic liver disease. Gastroenterology 1972;63:102–111
Wiest R, Shah V, Sessa WC, Groszmann RJ. NO overproduction by eNOS precedes hyperdynamic splanchnic circulation in portal hypertensive rats. Am J Physiol 1999;276:G1043–G1051
Iwakiri Y, Tsai MH, McCabe TJ, Gratton JP, Fulton D, Groszmann RJ, et al. Phosphorylation of eNOS initiates excessive NO production in early phases of portal hypertension. Am J Physiol Heart Circ Physiol 2002;282:H2084–H2090
Grace JA, Klein S, Herath CB, Granzow M, Schierwagen R, Masing N, et al. Activation of the MAS receptor by angiotensin-(1–7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology 2013;145(874–884):e875
Fernandez-Varo G, Ros J, Morales-Ruiz M, Cejudo-Martin P, Arroyo V, Sole M, et al. Nitric oxide synthase 3-dependent vascular remodeling and circulatory dysfunction in cirrhosis. Am J Pathol 2003;162:1985–1993
Heinemann A, Wachter CH, Fickert P, Trauner M, Stauber RE. Vasopressin reverses mesenteric hyperemia and vasoconstrictor hyporesponsiveness in anesthetized portal hypertensive rats. Hepatology 1998;28:646–654
Song D, Liu H, Sharkey KA, Lee SS. Hyperdynamic circulation in portal-hypertensive rats is dependent on central c-fos gene expression. Hepatology 2002;35:159–166
Moleda L, Trebicka J, Dietrich P, Gabele E, Hellerbrand C, Straub RH, et al. Amelioration of portal hypertension and the hyperdynamic circulatory syndrome in cirrhotic rats by neuropeptide Y via pronounced splanchnic vasoaction. Gut 2011;60:1122–1132
Ezkurdia N, Coll M, Raurell I, Rodriguez S, Cuenca S, Gonzalez A, et al. Blockage of the afferent sensitive pathway prevents sympathetic atrophy and hemodynamic alterations in rat portal hypertension. Liver Int 2012;32:1295–1305
Ezkurdia N, Raurell I, Rodriguez S, Gonzalez A, Esteban R, Genesca J, et al. Inhibition of neuronal apoptosis and axonal regression ameliorates sympathetic atrophy and hemodynamic alterations in portal hypertensive rats. PLoS ONE 2014;9:e84374
Dietrich P, Moleda L, Kees F, Muller M, Straub RH, Hellerbrand C, et al. Dysbalance in sympathetic neurotransmitter release and action in cirrhotic rats: impact of exogenous neuropeptide Y. J Hepatol 2013;58:254–261
Hartl J, Dietrich P, Moleda L, Muller-Schilling M, Wiest R. Neuropeptide Y restores non-receptor-mediated vasoconstrictive action in superior mesenteric arteries in portal hypertension. Liver Int 2015;35:2556–2563
Li TH, Huang CC, Yang YY, Lee KC, Hsieh SL, Hsieh YC, et al. Thalidomide improves the intestinal mucosal injury and suppresses mesenteric angiogenesis and vasodilatation by down-regulating inflammasomes-related cascades in cirrhotic rats. PLoS ONE 2016;11:e0147212
Fernandez-Varo G, Oro D, Cable EE, Reichenbach V, Carvajal S, de la Presa BG, et al. Vasopressin 1a receptor partial agonism increases sodium excretion and reduces portal hypertension and ascites in cirrhotic rats. Hepatology 2016;63:207–216
Yada A, Iimuro Y, Uyama N, Uda Y, Okada T, Fujimoto J. Splenectomy attenuates murine liver fibrosis with hypersplenism stimulating hepatic accumulation of Ly-6C(lo) macrophages. J Hepatol 2015;63:905–916
Kajita M, Murata T, Horiguchi K, Iizuka M, Hori M, Ozaki H. iNOS expression in vascular resident macrophages contributes to circulatory dysfunction of splanchnic vascular smooth muscle contractions in portal hypertensive rats. Am J Physiol Heart Circ Physiol 2011;300:H1021–H1031
Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol 2016;13:347–353
Caldwell S, Lisman T. The cirrhotic platelet: shedding light on an enigma. Hepatology 2017;65:407–410
Coller BS. The platelet: life on the razor’s edge between hemorrhage and thrombosis. Transfusion 2014;54:2137–2146
Iwakiri Y, Grisham M, Shah V. Vascular biology and pathobiology of the liver: report of a single-topic symposium. Hepatology 2008;47:1754–1763
Kawasaki T, Murata S, Takahashi K, Nozaki R, Ohshiro Y, Ikeda N, et al. Activation of human liver sinusoidal endothelial cell by human platelets induces hepatocyte proliferation. J Hepatol 2010;53:648–654
Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep 2012;2:1684–1696
Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun 2017;8:14361
Chung C, Iwakiri Y. The lymphatic vascular system in liver diseases: its role in ascites formation. Clin Mol Hepatol 2013;19:99–104
Iwakiri Y. The lymphatic system: a new frontier in hepatology. Hepatology 2016;64:706–707
Tanaka M, Iwakiri Y. The hepatic lymphatic vascular system: structure, function, markers, and lymphangiogenesis. Cell Mol Gastroenterol Hepatol 2016;2:733–749
Rautou PE, Tatsumi K, Antoniak S, Owens AP 3rd, Sparkenbaugh E, Holle LA, et al. Hepatocyte tissue factor contributes to the hypercoagulable state in a mouse model of chronic liver injury. J Hepatol 2016;64:53–59
Gonzalez-Reimers E, Quintero-Platt G, Martin-Gonzalez C, Perez-Hernandez O, Romero-Acevedo L, Santolaria-Fernandez F. Thrombin activation and liver inflammation in advanced hepatitis C virus infection. World J Gastroenterol 2016;22:4427–4437
Fiorucci S, Antonelli E, Distrutti E, Severino B, Fiorentina R, Baldoni M, et al. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology 2004;39:365–375
Violi F, Basili S, Raparelli V, Chowdary P, Gatt A, Burroughs AK. Patients with liver cirrhosis suffer from primary haemostatic defects? Fact or fiction? J Hepatol 2011;55:1415–1427
Lalor PF, Herbert J, Bicknell R, Adams DH. Hepatic sinusoidal endothelium avidly binds platelets in an integrin-dependent manner, leading to platelet and endothelial activation and leukocyte recruitment. Am J Physiol Gastrointest Liver Physiol 2013;304:G469–G478
Yoshida S, Ikenaga N, Liu SB, Peng ZW, Chung J, Sverdlov DY, et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014;147:1378–1392
Funding
This work was supported by NIH grants R21AA023599 and Connecticut DPH grant#2015-0901 (Y.I.) and NIH grant T32 007356 (M.M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Yasuko Iwakiri and Matthew McConnell declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Not applicable.
Rights and permissions
About this article

Cite this article
McConnell, M., Iwakiri, Y. Biology of portal hypertension. Hepatol Int 12 (Suppl 1), 11–23 (2018). https://doi.org/10.1007/s12072-017-9826-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-017-9826-x