
Abstract
Specific variations in the amino acid sequence of prion protein (PrP) are key determinants of susceptibility to prion diseases. We previously showed that an amino acid substitution specific to canids confers resistance to prion diseases when expressed in mice and demonstrated its dominant-negative protective effect against a variety of infectious prion strains of different origins and characteristics. Here, we show that expression of this single amino acid change significantly increases survival time in transgenic mice expressing bank vole cellular prion protein (PrPC), which is inherently prone to misfolding, following inoculation with two distinct prion strains (the CWD-vole strain and an atypical strain of spontaneous origin). This amino acid substitution hinders the propagation of both prion strains, even when expressed in the context of a PrPC uniquely susceptible to a wide range of prion isolates. Non-inoculated mice expressing this substitution experience spontaneous prion formation, but showing an increase in survival time comparable to that observed in mutant mice inoculated with the atypical strain. Our results underscore the importance of this PrP variant in the search for molecules with therapeutic potential against prion diseases.




Similar content being viewed by others

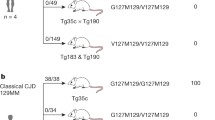
References
Colby DW, Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3(1):a006833. https://doi.org/10.1101/cshperspect.a006833
Collins SJ, Lawson VA, Masters CL (2004) Transmissible spongiform encephalopathies. Lancet 363(9402):51–61
Prusiner SB (1998) The prion diseases. Brain Pathol 8(3):499–513
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136–144
Stewart P, Campbell L, Skogtvedt S, Griffin KA, Arnemo JM, Tryland M, Girling S, Miller MW et al (2012) Genetic predictions of prion disease susceptibility in carnivore species based on variability of the prion gene coding region. PLoS One 7(12):e50623. https://doi.org/10.1371/journal.pone.0050623
Fernandez-Borges N, Parra B, Vidal E, Erana H, Sanchez-Martin MA, de Castro J, Elezgarai SR, Pumarola M et al (2017) Unraveling the key to the resistance of canids to prion diseases. PLoS Pathog 13(11):e1006716. https://doi.org/10.1371/journal.ppat.1006716
Vidal E, Fernandez-Borges N, Pintado B, Ordonez M, Marquez M, Fondevila D, Torres JM, Pumarola M et al (2013) Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci 33(18):7778–7786. https://doi.org/10.1523/JNEUROSCI.0244-13.2013
Polymenidou M, Trusheim H, Stallmach L, Moos R, Julius C, Miele G, Lenz-Bauer C, Aguzzi A (2008) Canine MDCK cell lines are refractory to infection with human and mouse prions. Vaccine 26(21):2601–2614. https://doi.org/10.1016/j.vaccine.2008.03.035
Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, Chakrabartty A (2010) Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A 107(46):19808–19813. https://doi.org/10.1073/pnas.1005267107
Chianini F, Fernandez-Borges N, Vidal E, Gibbard L, Pintado B, de Castro J, Priola SA, Hamilton S et al (2012) Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A 109(13):5080–5085. https://doi.org/10.1073/pnas.1120076109
Bian J, Khaychuk V, Angers RC, Fernandez-Borges N, Vidal E, Meyerett-Reid C, Kim S, Calvi CL et al (2017) Prion replication without host adaptation during interspecies transmissions. Proc Natl Acad Sci U S A 114(5):1141–1146. https://doi.org/10.1073/pnas.1611891114
Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, Cartoni C, Ingrosso L et al (2006) Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2(2):e12. https://doi.org/10.1371/journal.ppat.0020012
Agrimi U, Nonno R, Dell'Omo G, Di Bari MA, Conte M, Chiappini B, Esposito E, Di Guardo G et al (2008) Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog 4(7):e1000113. https://doi.org/10.1371/journal.ppat.1000113
Pirisinu L, Di Bari MA, D'Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS et al (2016) Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443. https://doi.org/10.1038/srep20443
Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10(4):e1003990. https://doi.org/10.1371/journal.ppat.1003990
Di Bari MA, Nonno R, Castilla J, D'Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J et al (2013) Chronic wasting disease in bank voles: Characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 9(3):e1003219. https://doi.org/10.1371/journal.ppat.1003219
Cartoni C, Schinina ME, Maras B, Nonno R, Vaccari G, Di Baria MA, Conte M, Liu QG et al (2005) Identification of the pathological prion protein allotypes in scrapie-infected heterozygous bank voles (Clethrionomys glareolus) by high-performance liquid chromatography-mass spectrometry. J Chromatogr A 1081(1):122–126
Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ et al (2012) Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc Natl Acad Sci U S A 109(9):3498–3503. https://doi.org/10.1073/pnas.1121556109
Will RG, Ironside JW (2017) Sporadic and infectious human prion diseases. Cold Spring Harb Perspect Med 7(1). https://doi.org/10.1101/cshperspect.a024364
Otero A, Bolea R, Hedman C, Fernández-Borges N, Marín B, López-Pérez O, Barrio T, Eraña H et al (2017) An amino acid substitution found in animals with low susceptibility to prion diseases confers a protective dominant-negative effect in prion-infected transgenic mice. Mol Neurobiol 55:6182–6192. https://doi.org/10.1007/s12035-017-0832-8
Castilla J, Gutierrez-Adan A, Brun A, Doyle D, Pintado B, Ramirez MA, Salguero FJ, Parra B et al (2004) Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci 24(21):5063–5069. https://doi.org/10.1523/Jneurosci.5400-03.2004
Fernandez-Borges N, Di Bari MA, Erana H, Sanchez-Martin M, Pirisinu L, Parra B, Elezgarai SR, Vanni I et al (2018) Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol 135(2):179–199. https://doi.org/10.1007/s00401-017-1782-y
Fraser H, Dickinson AG (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78(3):301–311
Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA (2000) The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol 156(1):51–56. https://doi.org/10.1016/S0002-9440(10)64705-0
Andreoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, Lugan S, Corbiere F et al (2004) PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med 10(6):591–593. https://doi.org/10.1038/nm1055
Monleon E, Monzon M, Hortells P, Bolea R, Acin C, Vargas F, Badiola JJ (2005) Approaches to scrapie diagnosis by applying immunohistochemistry and rapid tests on central nervous and lymphoreticular systems. J Virol Methods 125(2):165–171. https://doi.org/10.1016/j.jviromet.2005.01.013
Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121(2):195–206. https://doi.org/10.1016/j.cell.2005.02.011
Pirisinu L, Marcon S, Di Bari MA, D'Agostino C, Agrimi U, Nonno R (2013) Biochemical characterization of prion strains in bank voles. Pathogens 2(3):446–456. https://doi.org/10.3390/pathogens2030446
Westaway D, Zuliani V, Cooper CM, Da Costa M, Neuman S, Jenny AL, Detwiler L, Prusiner SB (1994) Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev 8(8):959–969
Clouscard C, Beaudry P, Elsen JM, Milan D, Dussaucy M, Bounneau C, Schelcher F, Chatelain J et al (1995) Different allelic effects of the codons 136 and 171 of the prion protein gene in sheep with natural scrapie. J Gen Virol 76(Pt 8):2097–2101
Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB (1997) Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 94(19):10069–10074
Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, Prusiner SB (2000) Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol 74(9):4351–4360
Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond SJ, Cohen FE, Prusiner SB et al (2002) Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci U S A 99(20):13079–13084. https://doi.org/10.1073/pnas.182425299
Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S (2009) Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog 5(7):e1000535. https://doi.org/10.1371/journal.ppat.1000535
Palmer MS, Dryden AJ, Hughes JT, Collinge J (1991) Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 352(6333):340–342. https://doi.org/10.1038/352340a0
Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T (1998) Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol 43(6):826–828. https://doi.org/10.1002/ana.410430618
Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S et al (2015) A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522(7557):478–481. https://doi.org/10.1038/nature14510
Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J (2000) Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci U S A 97(18):10248–10253
Collinge J (2001) Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci 24:519–550. https://doi.org/10.1146/annurev.neuro.24.1.519
Atarashi R, Sim VL, Nishida N, Caughey B, Katamine S (2006) Prion strain-dependent differences in conversion of mutant prion proteins in cell culture. J Virol 80(16):7854–7862. https://doi.org/10.1128/JVI.00424-06
Striebel JF, Race B, Meade-White KD, LaCasse R, Chesebro B (2011) Strain specific resistance to murine scrapie associated with a naturally occurring human prion protein polymorphism at residue 171. PLoS Pathog 7(9):e1002275. https://doi.org/10.1371/journal.ppat.1002275
Houston F, Goldmann W, Chong A, Jeffrey M, Gonzalez L, Foster J, Parnham D, Hunter N (2003) Prion diseases: BSE in sheep bred for resistance to infection. Nature 423(6939):498. https://doi.org/10.1038/423498a
Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, Hamilton S, Finlayson J et al (2008) The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol 89(Pt 12):2975–2985. https://doi.org/10.1099/vir.0.2008/005520-0
Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, Jakubcova T, Hamdan S et al (2013) Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog 9(9):e1003643. https://doi.org/10.1371/journal.ppat.1003643
Fernandez-Borges N, Erana H, Elezgarai SR, Harrathi C, Gayosso M, Castilla J (2013) Infectivity versus seeding in neurodegenerative diseases sharing a prion-like mechanism. Int J Cell Biol 2013:583498. https://doi.org/10.1155/2013/583498
Prusiner SB, Scott M, Foster D, Pan KM, Groth D, Mirenda C, Torchia M, Yang SL et al (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63(4):673–686
Hope J, Morton LJ, Farquhar CF, Multhaup G, Beyreuther K, Kimberlin RH (1986) The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J 5(10):2591–2597
Priola SA, Caughey B, Race RE, Chesebro B (1994) Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol 68(8):4873–4878
Jahandideh S, Jamalan M, Faridounnia M (2015) Molecular dynamics study of the dominant-negative E219K polymorphism in human prion protein. J Biomol Struct Dyn 33(6):1315–1325. https://doi.org/10.1080/07391102.2014.945486
Horiuchi M, Priola SA, Chabry J, Caughey B (2000) Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A 97(11):5836–5841. https://doi.org/10.1073/pnas.110523897
Seelig DM, Goodman PA, Skinner PJ (2016) Potential approaches for heterologous prion protein treatment of prion diseases. Prion 10(1):18–24. https://doi.org/10.1080/19336896.2015.1123372
Lee CI, Yang Q, Perrier V, Baskakov IV (2007) The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci 16(10):2166–2173. https://doi.org/10.1110/ps.072954607
Zhou S, Shi D, Liu X, Liu H, Yao X (2016) Protective V127 prion variant prevents prion disease by interrupting the formation of dimer and fibril from molecular dynamics simulations. Sci Rep 6:21804. https://doi.org/10.1038/srep21804
Acknowledgments
We thank MINECO for the Severo Ochoa Excellence Accreditation (SEV-2016-0644). The authors would like to thank the following for their support: the IKERBasque Foundation, the staff at the CIC bioGUNE animal facility and Patricia Piñeiro and Dr. Jan Langeveld from Central Veterinary Institute, Wageningen for kindly providing the 12B2 antibody.
Funding
This work was supported financially by the Spanish (AGL2015-65046-C2-1-R, AGL2015-65560-R) (MINECO/FEDER) and Interreg (POCTEFA EFA148/16) grants. Alicia Otero was supported by a research grant from the Government of Aragón (C020/2014) co-financed by the European Social Fund.
Author information
Authors and Affiliations
Contributions
JC and RB conceived the study; AO, CH, NFB, HE, BM, and MM performed most of the experiments; MASM and RN collaborated in the creation of the transgenic lines used; AO, HE, JJB, RB, and JC evaluated the results; AO, RB, JJB, HE, and JC wrote and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics Statement
All procedures involving animals were approved by the University of Zaragoza’s Ethics Committee for Animal Experiments (permit number PI32/13) and were performed in accordance with recommendations for the care and use of experimental animals and with Spanish law (R.D. 1201/05).
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1080 kb)
Rights and permissions
About this article

Cite this article
Otero, A., Hedman, C., Fernández-Borges, N. et al. A Single Amino Acid Substitution, Found in Mammals with Low Susceptibility to Prion Diseases, Delays Propagation of Two Prion Strains in Highly Susceptible Transgenic Mouse Models. Mol Neurobiol 56, 6501–6511 (2019). https://doi.org/10.1007/s12035-019-1535-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1535-0