
Abstract
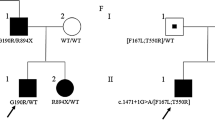
Myotonia congenita is a genetic disease caused by mutations in the CLCN1 gene, which encodes for the major chloride skeletal channel ClC-1, involved in the normal repolarization of muscle action potentials and consequent relaxation of the muscle after contraction. Two allelic forms are recognized, depending on the phenotype and the inheritance pattern: the autosomal dominant Thomsen disease with milder symptoms and the autosomal recessive Becker disorder with a severe phenotype. Before the recent advances of molecular testing, the diagnosis and genetic counseling of families was a challenge due to the large number of mutations in the CLCN1 gene, found both in homozygous or in heterozygous state. Here, we studied a consanguineous family in which three members presented a variable phenotype of myotonia, associated to a combination of three different mutations in the CLCN1 gene. A pathogenic splicing site mutation which causes the skipping of exon 17 was present in homozygosis in one very severely affected son. This mutation was present in compound heterozygosis in the consanguineous parents, but interestingly it was associated to a different second variant in the other allele: c.1453 A > G in the mother and c.1842 G > C in the father. Both displayed variable, but less severe phenotypes than their homozygous son. These results highlight the importance of analyzing the combination of different variants in the same gene in particular in families with patients displaying different phenotypes. This approach may improve the diagnosis, prognosis, and genetic counseling of the involved families.


Similar content being viewed by others
Availability of Data and Material
All data generated and/or analyzed during the study are available upon request.
References
Brugnoni R, Kapetis D, Imbrici P et al (2013) A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet 58:581–587. https://doi.org/10.1038/jhg.2013.58
Chen L, Schaerer M, Lu ZH et al (2004) Exon 17 skipping inCLCN1 leads to recessive myotonia congenita. Muscle Nerve 29:670–676. https://doi.org/10.1002/mus.20005
Cherian A, Baheti N, Kuruvilla A (2008) Muscle channelopathies and electrophysiological approach. Ann Indian Acad Neurol 11:20. https://doi.org/10.4103/0972-2327.40221
Colding-Jørgensen E, DunØ M, Schwartz M, Vissing J (2003) Decrement of compound muscle action potential is related to mutation type in myotonia congenita. Muscle Nerve 27:449–455. https://doi.org/10.1002/mus.10347
Colding-Jørgensen E (2004) Thomsens sygdom (myotonia congenita) [Thomsen disease (myotonia congenita)]. Ugeskr Laeger 166:3179–3184
Dubowitz V, Sewry CA, Oldfors A (2013) Muscle biopsy: a practical approach, 4th Edtion. Elsevier
Dupré N, Chrestian N, Bouchard J-P et al (2009) Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord 19:330–334. https://doi.org/10.1016/j.nmd.2008.01.007
Fialho D, Schorge S, Pucovska U et al (2007) Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain 130:3265–3274. https://doi.org/10.1093/brain/awm248
Hoche F, Seidel K, Barbosa-Sicard E et al (2014) Novel N-terminal truncating CLCN1 mutation in severe becker disease. Muscle Nerve 50:866–867. https://doi.org/10.1002/mus.24312
Imbrici P, Altamura C, Pessia M et al (2015) ClC-1 chloride channels: state-of-the-art research and future challenges. Front Cell Neurosci 09. https://doi.org/10.3389/fncel.2015.00156
Kubisch C, Schmidt-Rose T, Fontaine B et al (1998) ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet 7:1753–1760. https://doi.org/10.1093/hmg/7.11.1753
Lehmann-Horn F, Jurkat-Rott K (1999) Voltage-gated ion channels and hereditary disease. Physiol Rev 79:1317–1372. https://doi.org/10.1152/physrev.1999.79.4.1317
Mazón MJ, Barros F, De la Peña P et al (2012) Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul Disord 22:231–243. https://doi.org/10.1016/j.nmd.2011.10.013
Meyer-Kleine C, Steinmeyer K, Ricker K et al (1995) Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCNI) leading to myotonia. Am J Hum Genet 57:1325
Orsini C, Petillo R, D’ Ambrosio P et al (2020) CLCN1 molecular characterization in 19 South-Italian patients with dominant and recessive type of myotonia congenita. Front Neurol 11. https://doi.org/10.3389/fneur.2020.00063
Pusch M (2002) Myotonia caused by mutations in the muscle chloride channel geneCLCN1. Hum Mutat 19:423–434. https://doi.org/10.1002/humu.10063
Skálová D, Zídková J, Voháňka S et al (2013) CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS ONE 8. https://doi.org/10.1371/journal.pone.0082549
Steinmeyer K, Klocke R, Ortland C et al (1991) Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354:304–308. https://doi.org/10.1038/354304a0
Tan SV, Z’Graggen WJ, Boërio D et al (2014) Chloride channels in myotonia congenita assessed by velocity recovery cycles. Muscle Nerve 49:845–857. https://doi.org/10.1002/mus.24069
Trip J, Drost G, Verbove DJ et al (2008) In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur J Hum Genet 16:921–929. https://doi.org/10.1038/ejhg.2008.39
Wollnik B, Kubisch C, Steinmeyer K, Pusch M (1997) Identification of functionally important regions of the muscular chloride channel ClC-1 by analysis of recessive and dominant myotonic mutations. Hum Mol Genet 6:805–811. https://doi.org/10.1093/hmg/6.5.805
Wu F-F, Ryan A, Devaney J et al (2002) Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 125:2392–2407. https://doi.org/10.1093/brain/awf246
Acknowledgments
We would like to thank Fundação de Amparo à Pesquisa do Estado de São Paulo—Centro de Pesquisa, Inovação e Difusão (FAPESP-CEPID), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Instituto Nacional de Ciência e Tecnologia (INCT), FINEP, for the financial support. We would also like to thank the technical support of the following researchers: Adriano S. Senkevics, Dinorah Zilbersztajn-Gotlieb, Lydia U. Yamamoto, Viviane P. Muniz, and Leticia Nogueira. We are also very grateful to Dr. Paula Onofre-Oliveira for the English correction and improvement.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo—Centro de Pesquisa, Inovação e Difusão (FAPESP-CEPID), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Instituto Nacional de Ciência e Tecnologia (INCT), FINEP.
Author information
Authors and Affiliations
Contributions
Lucas Santos Souza conceived, designed, and performed the analysis and elaborated, wrote, and reviewed the manuscript. Priscila Calyjur performed the analysis. Antonio Fernando Ribeiro Jr performed the muscle protein analysis. Juliana Gurgel-Giannetti performed the medical and/or physical evaluation of the patients, and contributed with the manuscript revision. Rita Cassia Mingroni Pavanello performed the medical and/or physical evaluation of the patients. Mayana Zatz performed the genetic counseling and contributed with the manuscript revision. Mariz Vainzof conceived, designed, and performed the analysis and elaborated, wrote, and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
This work is in accordance and was approved by the ethics committee of the Biosciences Institute of the University of Sao Paulo, and the DNA samples are deposited in the biobank repository of the Human Genome and Stem Cells Research Center of IB-USP.
Consent to Participate
All subjects participating in this study signed an appropriated informed consent.
Consent for Publication
All subjects participating in this study signed an appropriated informed consent for publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Souza, L.S., Calyjur, P., Ribeiro, A.F. et al. Association of Three Different Mutations in the CLCN1 Gene Modulating the Phenotype in a Consanguineous Family with Myotonia Congenita. J Mol Neurosci 71, 2275–2280 (2021). https://doi.org/10.1007/s12031-020-01785-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01785-4