
Abstract
An enriched environment (EE) can stimulate the recovery of neurological function following a cerebral ischaemia-reperfusion injury; however, the impact of EE’s on mitochondrial function has been insufficiently studied. Our research aimed to assess whether EE’s therapeutic impact involved the enhancement of mitochondrial dysfunction. Following 2 weeks of EE training, we tested both mitochondrial function and mitochondria-associated protein expression within the cerebral cortex following cerebral ischaemia-reperfusion injury. We subjected Sprague-Dawley rats to transient focal cerebral ischaemia and categorized the rats into three separate groups, i.e. an enriched environment (EE) group, a standard condition (SC) group and a sham control group (no middle cerebral artery embolization). The rats within the EE group were raised in enriched conditions for 2 weeks, while the rats within the SC group, in comparison, were reared in standard conditions for 2 weeks. After 2 weeks, the cerebral cortices of the rats were removed. We then measured a series of indices, i.e. the protein expression of peroxisome proliferator-activated receptor gamma coactivator (PGC-1α), nuclear respiratory factor-1 (NRF-1), mitochondrial transcription factor A (TFAM) and mitochondrial protein cytochrome C oxidase subunit IV (COX IV). Furthermore, the number of mitochondria was evaluated through electron microscopy.EE upregulated the protein expression of PGC-1α, NRF-1 as well as TFAM, which function as the master regulators of mitochondrial biogenesis, in comparison with the SC group. The EE group’s COX IV protein expression also exhibited an increase. Moreover, the amount of mitochondria in the peri-infarct region of the cortex increased as result of EE training. Over 2 weeks, EE training significantly increased mitochondrial biogenesis-associated protein expression and mitochondrial function. A possible mechanism of the EE leading to the improvement of neurological function is that it increases brain mitochondrial biogenesis after the rats’ cerebral ischaemia-reperfusion injury. Mitochondrial biogenesis stimulation or enhancement could become an innovative strategy for neuroprotection in future treatment.
Similar content being viewed by others


Introduction
The primary cause of adult intellectual/physical dysfunction as well as mortality around the globe is stroke. EE could induce angiogenesis (Yu et al. 2014; Zhang et al. 2017), synaptic neogenesis (Bayat et al. 2015) and neurogenesis (Kuptsova et al. 2015), which are beneficial in improving neurological dysfunction after cerebral ischaemia-reperfusion injury. Mitochondria primarily work to generate cellular energy in the form of ATP through oxidative phosphorylation by the mitochondrial electron transport chain (ETC) and power the processes that are regarded as neuroplasticity through the supply of ATP. It has been demonstrated that mitochondrial dysfunction exacerbates neuronal injury following cerebral ischaemia due to the profound reliance of the nerve cells upon the mitochondria in meeting their high energy demand (Jones et al. 2012). Moreover, it is widely understood that mitochondrial homeostasis plays a significant role in a broad array of cellular functions and signalling events. Accordingly, promoting mitochondrial biogenesis or the repair of mitochondrial function after cerebral ischaemia is of high importance.
Some transcriptional regulators are involved in the process of mitochondrial biogenesis. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α), integrating physiological signals, is essential to regulating and enhancing mitochondrial biogenesis (Kleiner et al. 2009). PGC-1α leads to mitochondrial biogenesis through its activation of a host of transcription factors, i.e. nuclear respiratory factor 1 (NRF1) and nuclear respiratory factor 2 (NRF2), which can activate mitochondrial transcription factor A (TFAM).
The existing literature shows that ischaemic or hypoxic injury increases mitochondrial biogenesis (Bai et al. 2017; Xie et al. 2014). It has been shown that the process of mitochondrial biogenesis could be induced by exercise following brain ischaemia in rats. Nevertheless, the existing evidence has not clearly explained the process of how an EE triggers mitochondrial biogenesis and function.
We studied whether EE exerts an impact on cerebral mitochondrial biogenesis through the investigation of an ischaemic rat model. It is hypothesized that mitochondrial biogenesis regulation could help improve brain recovery from ischaemic injury. Cortical expression of mitochondria-specific transcription factors, protein levels and the mitochondria count after ischaemic injury triggered by transient middle cerebral artery occlusion (tMCAO)-induced were also analysed in this study.
Materials and Methods
Schematic Illustration of the Experimental Design
As we can see from Fig. 1, all points in time for subsequent operations and tests are described.

is a schematic diagram of the experimental design. Evaluations of Neurological Status were given on days 8 and 15, and Western blots and mitochondrial function were detected on day 15 after tMCAO. Abbreviations: tMCAO, transient Middle Cerebral Artery Occlusion; EE, Enriched Environment; SC, Standard housing Conditions; PGC-1α, Peroxisome proliferator-activated receptor coactivator-1α; NRF, Nuclear Respiratory Factor 1; TFAM, Mitochondrial Transcription Factor A; and COX IV, Cytochrome C oxidase subunit IV
Animals and the Ischaemic Model
Approval for the experimental protocol was obtained from the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and a licence was obtained from the Ethical Committee of Fudan University, Shanghai, China (Permit Number: 201602170860), respectively. Through slightly modified left transient middle cerebral artery occlusion (tMCAO), ischaemia was developed in adult male Sprague-Dawley rats (SLAC Inc., Shanghai, China) weighing between 250 and 280 g (Yu et al. 2013a). Isoflurane anaesthesia was induced at 5% in advance of the tMCAO surgery and then kept between 1.5 and 2% throughout the surgery. The rats’ body temperature was kept at 37 °C for the duration of the whole process through the utilization of a circulating heating pad. A silicone tipped 4-0 surgical nylon monofilament was moved from the left external carotid artery into the left internal carotid artery’s lumen. Rats within the sham control group went through identical surgery but without MCAO. Through the employment of a laser Doppler flow metre, the occlusion surgical operation was considered successful if the surface cerebral blood flow (CBF) declined to 20% of baseline CBF (Moor Instruments, Devon, UK). The extraction of the filament was performed to allow reperfusion following 90 min of cerebral ischaemia.
All rats were kept within a space with controlled temperature (21 ± 2 °C, 60–70% humidity) and were given unlimited access to food and water in a 12-h light/dark cycle (light from 8:00 AM–8:00 PM; dark from 8:00 PM–8:00 AM). Maximum effort was made to minimize the number of experimental rats, as well as their suffering for the duration of the experiment.
Experiment groups and training
On the next day of tMCAO, neurological scores were calculated using a four-point scale (Yu et al. 2014). The experimental rats were considered to satisfy the criteria when they scored between one and three points and were grouped into EE and SC groups, respectively. As previously described (Jiang et al. 2016), rats within the EE group were raised in EE caged spaces (Patent number of the People’s Republic of China: ZL 2010 2 0560281.3) pictured in Fig. 2A. The EE caged space was 80 cm in width, 120 cm in length and 100 cm in height and was equipped with extra stimulating objects, i.e. climbable platforms, plastic tubes, tunnels, chains and toys, as well as small boxes. Moreover, the rats within the EE group were raised in the caged spaces in a group of 12 and were moved every 3 days to maintain the environmental novelty, which provided the rats with enhanced social stimulation. In comparison, the rats within the SC group were raised in a group of three in standard cages (30 cm in width, 40 cm in length and 20 cm in height) with no extra stimulating objects, as pictured in Fig. 2B.

EE and SC housing conditions
Evaluation of Neurological Status
Through the blinded utilization of a seven-point scale as used in the literature (Yu et al. 2013b), we evaluated the neurological status of the rats after 8 and 15 days. The scoring system is presented in Table 1.
Collection of Tissue Samples From the Cortex
Fifteen days after the occlusion surgery, the rats were euthanized to collect their ipsilateral cortical tissue around the penumbral area over a piece of gauze moistened with freezing 0.9% saline. The collected samples were then swiftly frozen and stored at −80°C using liquid nitrogen for later analysis. The peripheral penumbra of the infarct areas of the cortex was then processed into pieces 1 mm3 in size and stored in 2.5% glutaraldehyde for electron microscopic observation.
Electron Microscopy Assessment of the Number of Mitochondria
Six penumbral areas on every rat were selected on a random basis before being photographed at 6,500x magnification by transmission electron microscopy (TEM) and tallied based on the slightly modified abovementioned method (three animals per group) (Wu et al. 1999) in order to measure the number of mitochondria. Samples stored in 2.5% glutaraldehyde were cleansed with water three times with PBS and fixed with 1% osmium tetroxide for 1 h before being cleansed again with pure water, dehydrated using ethanol, embedded in #618 resin and stained with uranyl acetate and lead citrate. We prepared and examined ultrathin sections, employing a Reichert ultramicrotome and a CM120 electron microscope (Philips) at 80 kV.
Western Blot
The cortex proteins were extracted and measured in their entirety through BCA with the assistance of a protein assay kit. As described previously (Yu et al. 2014), a western blot was conducted. Protein (40 μg) was separated through the employment of 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes. It was found that the major antibodies were anti- PGC-1a(Abcam, UK, 1:1000), NRF-1(Abcam, UK, 1:1500) ,TFAM (Abcam, UK, 1:2000) and COX IV (Abcam, UK, 1:500). The loading control in this study was anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH: Abcam, UK, 1:2000). We then cleansed the membranes with water, after which they were incubated with secondary antibodies at room temperature for 1 h. We conducted the quantification of band intensity/optical density on scanned western blot images through the use of Image J software based on the blots of independent studies.
Measurement of Cellular ATP Levels
Through the utilization of an ATP assay kit based on the manufacturer’s instructions (Beyotime, China), we measured the cellular ATP levels in cerebral tissues. The homogenization of brain tissues was conducted in a freezing ATP-releasing buffer before proceeding to centrifugation at 12,000 g for 5 min. After the centrifugation, the supernatant was transferred to a new tube to facilitate ATP testing. We assayed luminescence from a 100 μl sample in a spectrophotometer (Bio-Rad) along with 100 μl of ATP detection buffer before normalizing it by protein concentration.
Statistical analysis
Statistical analysis was performed using SPSS 13.0 statistical software (SPSS Inc., USA). Through the combination of both a one-way analysis of variance and post hoc Fisher’s Protected Least Significant Difference (PLSD) tests, differences between the groups were analysed. Through a t-test of independent samples, the statistical significance of physiological variables was ascertained. P values smaller than 0.05 were considered statistically significant.
Result
Effect of EE on Neurological Status
No ischaemic area was found within the rats of the sham control group. Rats within the EE group exhibited greater enhancement in comparison to the rats within the SC group (p < 0.05) on day 8 and day 15, respectively, as illustrated in Fig. 3.

Neurological status. Rats within the EE group exhibited greater enhancement in comparison to rats within the SC group on postoperative day 8 and day 15. Data are expressed as mean ± SEM. *p < 0.05 versus the SC group, n = 6 for each group
Effect of EE on the Number of Mitochondria
The regulation of the number of mitochondria was reflected by the rates of mitochondrial biogenesis. Through electron microscopy measurement, we quantified the number of mitochondria in the cortex’s peri-infarct region. The number of mitochondria of the rats within the EE group, as illustrated in Fig. 4, was evidently higher than that of rats within the SC group.

Effects of EE on the number of mitochondria in the peri-infarct subfield of the cortex after ischaemic reperfusion. A, ultrastructure of mitochondria (red arrows) in the peri-infarct subfield of the cortex from the rats in the sham, EE and SC groups; B, quantification of the number of mitochondria per photomicrograph. Red arrows indicate mitochondria. The values are the mean ± SEM, n = 3 for each group and six photomicrographs were counted per animal. *p < 0.05 versus the SC group
Effect of EE on the Expression of Mitochondrial Biogenesis Factors
The transcription factors’ protein expression levels were measured in order to study the molecular process which could be the mechanism underlying a post-tMCAO EE-dependent increase in terms of mitochondrial biogenesis. It has been illustrated through the western blot analysis that the rearing of rats within the EE group greatly enhanced the protein levels of PGC-1a, NRF-1 and TFAM, which is shown in Fig. 5.

Western blot analysis of PGC-1a, NRF-1 and TFAM in the ischaemic boundary zone at 15 days following tMCAO. Relative to the SC group, rats in the EE group had significantly higher levels of PGC-1a, NRF-1 and TFAM. Optical density values normalized to their respective GAPDH loading control were mean ± SEM and graphed (relative expression). *p < 0.05 versus the SC group, n = 6 for each group
Effect of EE on the Expression of COX IV
Mitochondrial-specific proteins include mitochondrial respiratory protein cytochrome C oxidase subunit IV (COXIV). Our study on the expression of proteins boosts confidence in the hypothesis that mitochondrial biogenesis can be induced by an EE. The EE group rats’ COX IV protein levels witnessed a greater increase compared to that of the SC group rats, as illustrated in Fig. 6.

Western blot analysis of COX IV in the ischaemic boundary zone on day 15 after tMCAO. Relative to the SC group, rats in the EE group had significantly higher COX IV levels. Optical density values normalized to their respective GAPDH loading control were mean ± SEM and graphed (relative expression). *p < 0.05 versus the SC group, n = 6 for each group
Effect of EE on Mitochondrial Function
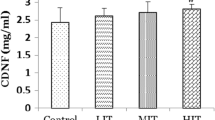
The attenuation of ischaemic brain injury relies critically on the protection of mitochondrial function. The ATP generation capacity was measured in order to assess the EE’s protective impact on mitochondrial function. As illustrated in Fig. 7, the cellular ATP levels of EE group rats exhibited a greater increase relative to that of SC group rats, implying that energy metabolism damaged by ischaemia can be improved by an EE.

Effect of the EE on mitochondrial function. The EE significantly improved mitochondrial function relative to SC. *p < 0.05 versus the SC group, n = 6 for each group
Correlation of PGC-1α Expression with Neurological Deficits
In the end, through the employment of Spearman’s correlation coefficient (R), PGC-1α protein expression and the neurological deficits were compared in order to evaluate a feasible correlation between the changes in both parameters. As illustrated in Fig. 8, the neurological status score negatively correlated with PGC-1α protein expression levels, i.e. higher PGC-1α protein expression levels were accompanied by lower neurological deficits.

Comparison of the correlation of PGC-1α with neurological deficits. R represents Spearman’s correlation coefficient. P < 0.05 was considered significant
Discussion
Brain ischaemia recovery can be facilitated by EE, as indicated by the literature (Xie et al. 2019). It has been demonstrated that neurologic function, induction of neurogenesis (Zhang et al. 2018) and angiogenesis (Zhang et al. 2017) can be improved by EE in particular. The protective impacts of EE are realized through the reduction of brain inflammation levels (Quattromani et al. 2014), moderation of the mitochondria of injured neurons (Chen et al. 2017) as well as the attenuation of blood brain barrier dysfunction (Diaz et al. 2016).
The primary endogenous protective process related to cerebral ischaemia is mitochondrial biogenesis, as evidenced by recent studies (Chen et al. 2010; Yin et al. 2008). Mitochondria were found by a host of studies to exert a major impact on the control of neuroplasticity (Habash et al. 2015). Neuroplasticity can be disturbed by mitochondrial dysfunction after stroke (Cheng et al. 2010; Raefsky and Mattson 2017; Sims and Anderson 2002). Mitochondria are essential cellular organelles in the sense that they, on the one hand, generate adenosine triphosphate (ATP) for cellular functions and, on the other hand, take part in many intracellular processes, e.g. oxidative response, regulation of cytosolic Ca2+ concentration, determination of cell death or survival, metabolic signalling pathways and cell division (Bai et al. 2017; Camara et al. 2011).
A subtle connection between mitochondrial biogenesis and neurological disorders was uncovered by recent studies (Mandemakers et al. 2007). Mitochondrial biogenesis was found to counteract the detrimental effects of oxidative stress (Habash et al. 2015) and provide sufficient energy in the neuroplasticity process that has been suggested as a novel target of the repair mechanism. The enhancement of mitochondrial biogenesis may reduce ischaemic brain injury (Valerio et al. 2011), and mitochondrial biogenesis is likely to minimize the effects of ischaemia-induced injury to the mitochondria by increasing their number. PGC-1α is commonly regarded as the master regulator and is often upregulated during times of increased energy demand (Anzell et al. 2018). In addition, the primary regulator of mitochondrial biogenesis (Wu et al. 1999) and the main inducer of the gene expression of NRF-1 (Valerio et al. 2011), which is situated at a distinctive connecting location of the TFAM gene (Scarpulla 2008) are both considered to be PGC-1α. The brain protein levels of TFAM and NRF-1 were measured, as both TFAM and NRF-1 were considered to take a critical role in both the transcription of mitochondrial encoded genes as well as the initiation of mtDNA replication, respectively (Campbell et al. 2012). The protein levels of both TFAM and NRF-1, mitochondrial function by EE training in I/R injury, ATP levels as well as the number of mitochondria can be increased substantially through EE treatment as evidenced by our study outcomes, which are also in line with existing literature (Kumari et al. 2012). Furthermore, it has been found that PGC-1α has an essential role, which might be connected with the neuronal requirement incurred by the rise in energy during the process, indicating that biogenesis takes a leading role in synapse engendering and preservation (Cheng et al. 2012) and neurological impairment recovery (Thornton et al. 2018).
As a mitochondria-specific protein, mitochondrial protein cytochrome C oxidase subunit IV (COX IV) can be detected within a high concentration of mitochondria (Borutaite et al. 2013). The fluctuation in COX IV levels was measured through examination of the cortex, which serves as an indirect mitochondrial biogenesis evaluation. After 14 days of EE rearing, the EE group rats’ cortical COX IV protein levels exhibited a greater increase than the SC group rats, which substantiates the proposition that an EE is conducive to mitochondrial biogenesis enhancement.
There were increases in the expression of PGC-1α, NRF-1, TFAM and COX IV following cerebral ischaemia-reperfusion injury. In addition, neurological status scores were found to be negatively correlated to the PGC-1α expression, i.e. when the expression of PGC-1α was higher, there was a better neurological status. EE-induced mitochondrial function and biogenesis in cortical neurons, as evidenced by our experiment outcomes, could be used as a powerful therapeutic measure in terms of brain plasticity enhancement following cerebral ischaemia-reperfusion injury, which, nevertheless, still requires further empirical studies in order for the precise process of the impact of an EE on mitochondria to be ascertained.
In summary, EE could, through mitochondrial biogenesis regulation, be beneficial in cerebral recovery from ischaemia-induced injury, as evidenced by our results indicating that EE is helpful in temporarily improving the expression of the mitochondrial factors leading to mitochondrial biogenesis following ischaemic brain injury. Additional studies to investigate the process controlling EE-induced mitochondrial biogenesis are required in order to elucidate this intricate EE-associated neuroprotection. Our study has provided evidence that mitochondrial biogenesis improvement and stimulation could become a new neuroprotective measure for future treatment.
References
Anzell AR, Maizy R, Przyklenk K, Sanderson TH (2018) Mitochondrial quality control and disease: insights into ischemia-reperfusion injury. Mol Neurobiol 55:2547–2564
Bai F, Guo F, Jiang T, Wei H, Zhou H, Yin H, Zhong H, Xiong L, Wang Q (2017) Arachidonyl-2-chloroethylamide alleviates cerebral ischemia injury Through Glycogen Synthase Kinase-3beta-Mediated Mitochondrial Biogenesis and Functional Improvement. Mol Neurobiol 54:1240–1253
Bayat M, Sharifi MD, Haghani M, Shabani M (2015) Enriched environment improves synaptic plasticity and cognitive deficiency in chronic cerebral hypoperfused rats. Brain Res Bull 119:34–40
Borutaite V, Toleikis A, Brown GC (2013) In the eye of the storm: mitochondrial damage during heart and brain ischaemia. FEBS J 280:4999–5014
Camara AK, Bienengraeber M, Stowe DF (2011) Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol 2:13
Campbell CT, Kolesar JE, Kaufman BA (2012) Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta 1819:921–929
Chen SD, Lin TK, Lin JW, Yang DI, Lee SY, Shaw FZ, Liou CW, Chuang YC (2010) Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 88:3144–3154
Chen X, Zhang X, Xue L, Hao C, Liao W, Wan Q (2017) Treatment with Enriched Environment Reduces Neuronal Apoptosis in the Periinfarct Cortex after Cerebral Ischemia/Reperfusion Injury. Cell Physiol Biochem 41:1445–1456
Cheng A, Hou Y, Mattson MP (2010) Mitochondria and neuroplasticity. ASN Neuro 2:e00045
Cheng A et al (2012) Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun 3:1250
Diaz R, Miguel PM, Deniz BF, Confortim HD, Barbosa S, Mendonça MCP, da Cruz-Höfling MA, Pereira LO (2016) Environmental enrichment attenuates the blood brain barrier dysfunction induced by the neonatal hypoxia-ischemia. Int J Dev Neurosci 53:35–45
Habash T, Saleh A, Roy Chowdhury SK, Smith DR, Fernyhough P (2015) The proinflammatory cytokine, interleukin-17A, augments mitochondrial function and neurite outgrowth of cultured adult sensory neurons derived from normal and diabetic rats. Exp Neurol 273:177–189
Jiang C, Yu K, Wu Y, Xie H, Liu G, Wu J, Jia J, Kuang S (2016) Enriched Environment Enhances Poststroke Neurological Function Recovery on Rat: Involvement of p-ERK1/2. J Stroke Cerebrovasc Dis 25:1590–1598
Jones AW, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G (2012) PGC-1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion. 12:86–99
Kleiner S, Nguyen-Tran V, Baré O, Huang X, Spiegelman B, Wu Z (2009) PPAR{delta} agonism activates fatty acid oxidation via PGC-1{alpha} but does not increase mitochondrial gene expression and function. J Biol Chem 284:18624–18633
Kumari S, Anderson L, Farmer S, Mehta SL, Li PA (2012) Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res 3:296–304
Kuptsova K, Kvist E, Nitzsche F, Jolkkonen J (2015) Combined enriched environment/atipamezole treatment transiently improves sensory functions in stroke rats independent from neurogenesis and angiogenesis. Romanian J Morphol Embryol 56:41–47
Mandemakers W, Morais VA, De Strooper B (2007) A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci 120:1707–1716
Quattromani MJ, Cordeau P, Ruscher K, Kriz J, Wieloch T (2014) Enriched housing down-regulates the Toll-like receptor 2 response in the mouse brain after experimental stroke. Neurobiol Dis 66:66–73
Raefsky SM, Mattson MP (2017) Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic Biol Med 102:203–216
Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638
Sims NR, Anderson MF (2002) Mitochondrial contributions to tissue damage in stroke. Neurochem Int 40:511–526
Thornton C, Jones A, Nair S, Aabdien A, Mallard C, Hagberg H (2018) Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett 592:812–830
Valerio A, Bertolotti P, Delbarba A, Perego C, Dossena M, Ragni M, Spano P, Carruba MO, de Simoni MG, Nisoli E (2011) Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J Neurochem 116:1148–1159
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 98:115–124
Xie Y et al (2014) Reperfusion promotes mitochondrial biogenesis following focal cerebral ischemia in rats. PLoS One 9:e92443
Xie H, Yu K, Zhou N, Shen X, Tian S, Zhang B, Wang Y, Wu J, Liu G, Jiang C, Hu R, Ayata C, Wu Y (2019) Enriched Environment Elicits Proangiogenic Mechanisms After Focal Cerebral Ischemia. Transl Stroke Res 10:150–159
Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J (2008) Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 39:3057–3063
Yu K, Wu Y, Hu Y, Zhang Q, Xie H, Liu G, Chen Y, Guo Z, Jia J (2013a) Neuroprotective effects of prior exposure to enriched environment on cerebral ischemia/reperfusion injury in rats: the possible molecular mechanism. Brain Res 1538:93–103
Yu K, Wu Y, Hu Y, Zhang Q, Xie H, Liu G, Chen Y, Guo Z, Jia J (2013b) Prior exposure to enriched environment reduces nitric oxide synthase after transient MCAO in rats. Neurotoxicology. 39:146–152
Yu K, Wu Y, Zhang Q, Xie H, Liu G, Guo Z, Li F, Jia J, Kuang S, Hu R (2014) Enriched environment induces angiogenesis and improves neural function outcomes in rat stroke model. J Neurol Sci 347:275–280
Zhang X, Chen XP, Lin JB, Xiong Y, Liao WJ, Wan Q (2017) Effect of enriched environment on angiogenesis and neurological functions in rats with focal cerebral ischemia. Brain Res 1655:176–185
Zhang Y, Xu D, Qi H, Yuan Y, Liu H, Yao S, Yuan S, Zhang J (2018) Enriched environment promotes post-stroke neurogenesis through NF-kappaB-mediated secretion of IL-17A from astrocytes. Brain Res 1687:20–31
Funding
This work was supported by Natural Science Foundation of China (NSFC, nos. 81672242, 81601961).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yu, K., Kuang, S., Wang, C. et al. Changes in Mitochondria-Associated Protein Expression and Mitochondrial Function in Response to 2 Weeks of Enriched Environment Training After Cerebral Ischaemia-Reperfusion Injury. J Mol Neurosci 70, 413–421 (2020). https://doi.org/10.1007/s12031-019-01428-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-019-01428-3