
Abstract
Mast cells (MCs) are critically involved in microbial defense by releasing antimicrobial peptides (such as cathelicidin LL-37 and defensins) and phagocytosis of microbes. In past years, it has become evident that in addition MCs may eliminate invading pathogens by ejection of web-like structures of DNA strands embedded with proteins known together as extracellular traps (ETs). Upon stimulation of resting MCs with various microorganisms, their products (including superantigens and toxins), or synthetic chemicals, MCs become activated and enter into a multistage process that includes disintegration of the nuclear membrane, release of chromatin into the cytoplasm, adhesion of cytoplasmic granules on the emerging DNA web, and ejection of the complex into the extracellular space. This so-called ETosis is often associated with cell death of the producing MC, and the type of stimulus potentially determines the ratio of surviving vs. killed MCs. Comparison of different microorganisms with specific elimination characteristics such as S pyogenes (eliminated by MCs only through extracellular mechanisms), S aureus (removed by phagocytosis), fungi, and parasites has revealed important aspects of MC extracellular trap (MCET) biology. Molecular studies identified that the formation of MCET depends on NADPH oxidase-generated reactive oxygen species (ROS). In this review, we summarize the present state-of-the-art on the biological relevance of MCETosis, and its underlying molecular and cellular mechanisms. We also provide an overview over the techniques used to study the structure and function of MCETs, including electron microscopy and fluorescence microscopy using specific monoclonal antibodies (mAbs) to detect MCET-associated proteins such as tryptase and histones, and cell-impermeant DNA dyes for labeling of extracellular DNA. Comparing the type and biofunction of further MCET decorating proteins with ETs produced by other immune cells may help provide a better insight into MCET biology in the pathogenesis of autoimmune and inflammatory disorders as well as microbial defense.
Similar content being viewed by others


Introduction
Formation of extracellular traps (ETs) by several types of leukocytes occurs as a late antimicrobial response to the presence of microbial invaders (in vivo) or special chemicals (mostly reported in in vitro experiences) [1, 2]. Although ET formation was primarily described as a mechanism used by leukocytes in microbial defense, ETs were later shown to be associated with several non-infectious pathologies including psoriasis, systemic lupus erythematosus (SLE), liver damage, acute pancreatitis, and cancer metastasis [2,3,4,5,6]. ETs, the thread-like complexes of decondensed DNA (nuclear or mitochondrial DNA [7]) with attached proteins from cytoplasmic granules, were first reported in neutrophils to act as an extracellular mechanism in microbial defense [8]. The formation of ETs in leukocytes results in the cell death of the leukocyte which from a molecular point of view is neither necrosis nor apoptosis [9]. Extracellular traps gained attention when they were reported to be produced by other myeloid cells such as monocytes [10] or eosinophils [11]. The molecular structure of ETs depends on the type of the producing cell and the stimuli; for instance, neutrophil ETs (NETs) are comprised of neutrophil elastase (NE), myeloperoxidase (MPO), cathepsin G, leukocyte proteinase 3 (PR3), lactoferrin, gelatinase, lysozyme C, calprotectin, cathelicidins, and defensins [9]. In contrast, mast cells (MCs), another innate immune cells, produce ETs (MCETs) containing histones, tryptase, and LL-37 [12] (Fig. 1a). The main biologic functions of these biomolecules and mediators are listed in Table 1. MCs are granulated leukocytes of innate immunity that differentiate in target tissues from CD117 + /CD34 + progenitors released from the bone marrow [13, 14]. Under the influence of growth factors such as stem cell factor (SCF), IL-3, IL-4, IL-9, IL-10, IL-33, and TGF-β [15], MC progenitors differentiate in functional mature cells that respond to a variety of environmental stimuli owing to expression of receptors including toll-like receptors and receptors to Fc portion of antibodies (such as FcεRI:IgE or FcγR: IgG) [16,17,18]. Beyond their classic role in allergic and anaphylactic reactions [19], MCs play an important role in microbial defense [12]. At very early steps of microbial invasion, MCs effectively recruit neutrophils to the site of infection by releasing TNF-α which is a preformed and stored mediator of MCs [20]. The results of experimental infection with S. aureus in MC-deficient KitW−sh/W−sh mice and corresponding wild type (WT) littermates or reconstitution of MC-deficient mice with MCs derived from WT mice showed that (a) In KitW−sh/W−sh mice recruitment of neutrophils and elimination of bacteria were impaired, (b) reconstituting the MC population in KitW−sh/W−sh mice by injection of MCs from WT mice could restore their ability to eliminate the bacteria, and (c) exogenous TNF-α could compensate the partial ineffectiveness of MC-deficient mice in recruiting neutrophils to the cite of infection supporting the notion that MC-released TNF-α participates actively in microbial defense [21] (Fig. 1b). MCs utilize both intracellular (including phagocytosis) and extracellular mechanisms (mainly via release of peptides with antimicrobial properties) for the elimination of invading pathogens [12, 22]. Additionally, MCs activate CD4+ T cells by acting as antigen presenting cells (APCs). It is now evident that MCs express MHC-II and costimulatory molecules such as OX40L, CD80, and CD86 to activate CD4+ T cells (expressing the corresponding receptors including OX-40 and CD28, respectively) and as such, orchestrate adaptive immune responses [23, 24]. Besides, MCs are abundant in B cell localizing areas in lymph nodes and the coculture of these two cell populations revealed that MCs induce the proliferation of both naïve and activated B cells and support their differentiation into IgA producing cells via expressing CD40L and releasing IL-6 [25]. Accordingly, MC-released IL-6 can play a critical role in the activation and proliferation of B cells in vivo [26]. MCs express different types of surface receptors to recognize microbes including TLR-2/Dectin-1 for the detection of C. albicans and produce nitric oxide (NO) which possesses cytotoxic effects against microorganisms [27, 28]. The ability of MCs to produce extracellular traps (ETs) was first reported in 2008 [12] (Fig. 2). ETosis of MCs and subsequent cell death can be inhibited by the NADPH oxidase inhibitor diphenyleneiodonium (DPI) indicating a critical role for reactive oxygen species (ROS) in MCET formation [12, 29]. In the following sections, we will review different aspects of MCETs with focus on their structure, microbial and chemical stimuli that induce their formation, their role in restriction of microbial infections, and finally possible involvement in several noninfectious pathologies [30]. Additionally, we will discuss the technical procedure commonly used to stain the different components of MCETs and visualizing them under microscope.

a structure of ETs, ET-associated proteins, and the nature of DNA depend on the producing cell types. b Role of MCs in antimicrobial defense against S. aureus: MCs release TNF-α which is a critical neutrophil attractant to the site of infection. MCs from MC-deficient KitW−sh/W−sh mice cannot effectively attract neutrophils when compared to the wild type Kit+/+ MCs. When WT MCs are injected to MC-deficient KitW−sh/W−sh mice, they restore their ability to eliminate the bacteria by recruiting neutrophils to the site of infection

Intracellular and extracellular mechanisms of microbial defense used by mast cells. (1) MCs act as antigen presenting cells by expressing MHC class II molecules and costimulatory molecules to activate CD4+ T cells and support the orchestration of adaptive immune responses; (2) MCs can act as phagocytes by directly engulfing invading pathogens and killing them in phagolysosomes; (3) MCs produce MCETs consisting of DNA, histones, LL-37, and tryptase to trap and immobilize invading pathogens; (4) MCs produce and release antimicrobial peptides such as the cathelicidin LL-37; (5) MCs effectively recruit other phagocytes to the site of infection by releasing cytokines such as TNF-α for neutrophil recruitment; and (6) MCs play a role in induction of proliferation in B cells by releasing cytokines and surface receptors
Cell Death Pathways in Innate Immune
There are four cell death pathways described in innate immune cells when they are exposed to special bacteria and viruses including non-lytic and silent cell death mainly apoptosis, and inflammatory programmed lytic types including necroptosis, pyroptosis, and ETosis [31].
Necroptosis
Engagement of TNF superfamily receptors, toll-like receptors (mainly TLR3 and TLR4), and interferon receptors drives the process of necroptosis during which the interaction between receptor-interacting protein kinase 1 (RIPK1) and RIPK3 leads in formation of heterodimer complex that promotes oligomerization of mixed-lineage kinase domain-like protein (MLKL)—acts as the RIPK3 substrate—through phosphorylation. MLKL oligomers translocate towards the plasma membrane and cause pore formation and further inflammatory response [32].
Pyroptosis
The canonical pathway of pyroptosis is initiated when inflammasome sensor proteins mainly NLRP3 recognize the K+ efflux induced by microbial pathogens, toxins, and DAMPs [33]. Inflammasomes activated by DAMPs and PAMPs bind to apoptosis-associated speck-like protein (ASC) and recruit procaspase-1 and activate caspase-1. The latter molecule cleaves proIL-18 /1β and mediates the cleavage of gasdermin D (GSDMD). The N-terminal fragment of GSDMD (GSDMD-NT) mediates the formation of the pores in the plasma membrane, through which IL-18 /1β are released and water influx occurs. The final consequences of these molecular events are cell swelling and finally osmotic lysis [34].
ETosis
In contrast to apoptosis, during ETosis, biologic changes such as nuclear condensation and DNA fragmentation do not happen. Indeed, nuclear chromatin decondensation in the cytoplasm is a common finding. Moreover, disintegration of the nucleus membrane, therefore cell death, results in release of nuclear DNA to form extracellular DNA nets [35]. From a molecular point of view, NADPH-oxidase-mediated production of ROS plays a key role in the formation of ETosis [36, 37]. Moreover, peptidyl arginine deiminase-mediated deimination of histone arginine residues to citrullines is another biochemical finding that contributes to chromatin decondensation [38]. Therefore, not interestingly hypercitrullinated histones are found in the structure of ETs when chemicals such as LPS and H2O2 act as the stimuli [39]. Since formation of ETs is followed by biologic changes including disintegration of the nuclear and cellular membranes, decondensation of chromatin and DNA structural modification mainly citrullination, and the release of both mitochondrial and nuclear DNA from the cells into the extracellular space, it is more likely that production of ETs results in the cell death [40] (Fig. 3).

The molecular basis of inflammatory programmed lytic cell death types including necroptosis, pyroptosis, and ETosis. Engagement of TNFR, TLR3 and TLR4, and interferon receptors drives the interaction between receptor-interacting protein kinase 1 (RIPK1) and RIPK3 that promotes oligomerization of mixed-lineage kinase domain-like protein (MLKL) MLKL oligomers cause pore formation. In pyroptosis, inflammasomes activated by DAMPs and PAMPs bind to apoptosis-associated speck-like protein (ASC) and recruit procaspase-1 and activate caspase-1. Then caspase-1 cleaves proIL-18/1β and gasdermin D (GSDMD). The N-terminal fragment of GSDMD (GSDMD-NT) mediates the formation of the pores in the plasma membrane, through which IL-18/1β are released and water influx occurs. During ETosis, decondensation of chromatin, histone citrullination, and release of DNA into cytoplasm occur. DNA ejects into the extracellular space along with NET-associated antimicrobial peptides
Extracellular Traps Formed by Immune Cells
Neutrophils
By studying inflammatory conditions including experimental shigellosis in rabbits and appendicitis in humans, Brinkmann and colleagues were the first to describe a novel extracellular anti-microbial mechanism in neutrophils in 2004. By staining histones, DNA, and neutrophil elastase, they reported the ability of neutrophils to eject DNA strands and utilize them for the trapping of pathogens [41]. A wide variety of stimuli including interferon (IFN)-α, interleukin (IL)-8, chemical agents (mainly phorbol myristate acetate; PMA), certain microbes, and their products have since been shown to induce the formation of neutrophil ETs (NETs) [42, 43]. NETosis is initiated by decondensation of chromatin, and the release of nuclear contents into the cytoplasm. In the final stage, DNA is released into the extracellular space to ensnare the invading pathogens [42]. Upon ejection of NETs, a variety of substances with bactericidal properties including proteases, LL-37, and protease-containing matrix metalloproteinase 9 (MMP-9) are released and contribute to the elimination of the pathogen [43]. Moreover, citrullinated histone H3 (H3Cit) and peptidyl arginine deiminase (PAD) are commonly released in conjunction with DNA [44] (Fig. 4a). NETosis is activated not only upon exposure to the above listed cytokines or chemicals but also the crosstalk of several cell types with neutrophils may induce the formation of NETs. Specifically, the production of NETs can be triggered by inorganic polyphosphate (polyP), notably also a secretory product of MCs which co-express it with CD68 [45]. The abundance of polyP expressing CD68+ MCs in the proximity of tumor cells in patients with colorectal cancer suggests that MCs may prime or trigger the production of NETs in cancer [45]. NETs have also been linked to procoagulant activity in patients with acute stroke. Indeed, the interaction between neutrophils and activated platelets induces the production and release of NETs decorated with phosphatidylserine (provides binding sites for the activation of coagulation factors when it is expressed on microvesicles or blood cells) [46]. Adhesion of coagulation factors and platelet-derived extracellular vesicles to NETs further contributes to the formation of thrombin and fibrin in stroke patients [46]. During NETosis, a variety of proteases are released from neutrophil granules and attach to DNA that have special biofunctions; for instance, neutrophil elastase, cathepsin G, and myeloperoxidase (MPO) are released from azurophilic (primary) granules, while lactoferrin and gelatinase are released from specific (secondary) granules and tertiary granules, respectively [41].

Production of ETs in neutrophils and eosinophils in response to chemical and biologic stimuli. a Stimuli including IL-8, IFN-α, PMA, and microbes induce the generation of NETs. Molecular events during ETosis include chromatin decondensation, DNA release into the cytoplasm, and release of DNA webs decorated with histones, LL-37, PAD, and MMP-9 into the extracellular space to trap invading pathogens. b Eosinophils produce EETs through releasing of mitochondrial DNA that becomes decorated with MBP upon exposure to stimuli including C5a, LPS, IgA/IgG, and GM-CSF/IL-5 + PAF
Eosinophils
Over the recent years, it has become evident that neutrophils are not the only myeloid cells able to produce ETs. Release of eosinophil ETs (EETs) was first demonstrated when blood purified eosinophils were primed by IL-5 or IFN-γ for 20 min and then exposed to lipopolysaccharide (LPS) or complement factor C5a. In contrast to NETosis, EETosis results from the ejection of mitochondrial rather than nuclear DNA, presumably in a ROS–dependent manner [11]. Immobilized IgA/IgG and GM-CSF/IL-5 with platelet-activating factor (PAF) are among other stimuli of EETosis in vitro [47]. Additionally, formation of EETs may be triggered by the presence of viral infection as eosinophils derived from Ovalbumin-sensitized BALB/cJ mice were shown to produce EETs following infection with respiratory syncytial virus (RSV) in vitro [48]. The released EETs were composed of DNA decorated with toxic major basic protein (MBP) [49] (Fig. 4b). Production of EETs has been primarily studied in the context of severe eosinophilic asthma. Eosinophils from patients with severe asthma were reported to be more activated than those with non-severe asthma, and these eosinophils produce higher levels of ROS and EETs. Notably, the number of EET producing eosinophils correlates negatively with forced expiratory volume in 1 s (FEV1) and the severity of the disease [50], indicating the potential functional relevance of EETs in asthma. Production of EETs in asthmatics was, however, not affected by allergen challenge or levels of eotaxin, IFN-γ, and IL-5 in bronchoalveolar lavage [49]. Investigations of the structure of EETs showed that eosinophils release Charcot-Leyden crystals (CLCs) during the formation of EETs. CLCs are composed of eosinophil protein galectin-10 and commonly found in patients with allergic diseases such as asthma [51]. Non-stimulated eosinophils or those treated with diphenyleneiodium chloride were rarely found to release the crystals showing that crystals were associated with the formation of EETs. Considering the fact that formation of many crystals usually is associated with tissue injury, more studies are needed to clarify the significance of Charcot-Leyden crystals released during EETosis [47].
Monocytes
Similar to eosinophils, monocytes can produce ETs by ejection of mitochondrial DNA that is decorated with global histones (H1, H2A/H2B, H3, H4) and citrullinated histones such as histone H4 citrullinated 3 (H4Cit3) [52]. Monocyte ETs (METs) can trap other cells, as demonstrated for spermatozoa from healthy individuals which showed a reduced mobility in the presence of monocytes simulated with E. coli [52]. Accordingly, monocytes have been found to be involved in microbial defense against parasites including viable Besnoitia besnoiti tachyzoites by the production of METs decorated with H3 histones and myeloperoxidase (MPO) [53]. In addition to humans, METosis has been reported in animals as well. In this regard, sensing of T. gondii-tachyzoites by monocytes of Harbour seals induces the formation of METs that results in entrapping and immobilizing of the parasite [54]. Notably, MET formation may also be induced by hormonal changes as demonstrated in a study of monocytes purified from peripheral blood of non-pregnant women during the menstrual cycle which showed that (a) more METs are produced during the luteal phase compared to the follicular phase and (b) revealed a positive correlation between the number of METs and serum levels of progesterone [55] (Fig. 5a).

Production of ETs in monocytes and macrophages: a and b ETs produced by monocytes and macrophages are decorated with different types of peptides and proteins. c Both forms of C. albicans, yeast cells, and hypae induce the release of METs from macrophages. Additionally, both live and heat-killed C. albicans induce the formation of METs; however, heat-killed C. albicans are more potent in triggering MET formation from macrophages due to the absence of their DNase activity
Macrophages
Both monocyte-derived macrophages and macrophage cell lines from humans and animals have been reported to produce and release macrophage extracellular traps (METs). Upon release, these web-like chromatin structures are decorated with H3Cit, granule proteases, and PAD similar to NETs [44]. Macrophages produce METs in response to a variety of microbes as well as to exotoxins of bacteria [56] (Fig. 5b) such as Mannheimia haemolytica (which causes bovine respiratory disease) and its leukotoxin or E. coli-derived hemolysin [56]. In macrophages stimulated with M. haemolytica, Aulik and colleagues showed that DNase treatment reduced the number of trapped and killed bacteria, thereby consolidating the role of METs in antimicrobial defense [56]. Consistently, Loureiro and coworkers demonstrated the role of METs in the control and killing of C. albicans in that (a) both forms of C. albicans yeast cells and hyphae can induce macrophages to produce METs and (b) both heat-killed and live C. albicans induce the generation of METs with the former more than the latter, possibly due to DNase activity in live C. albicans [57]. Importantly, this study introduces the DNase of C. albicans as a novel virulent factor. Indeed, METosis may act as a mechanism to confine the spread of C. albicans rather than killing the yeast [58] (Fig. 5c).
Mast Cell Extracellular Traps
Discovery and Early Reports
Just 4 years after the discovery of NETs by Brinkmann et al. [41], Köckritz-Blickwede and colleagues reported that just like neutrophils, MCs can produce extracellular traps [12]. Over the past decade, the structure, function, and relevance of MCETs in infectious disease have been elucidated by models of intracellular or extracellular bacterial infection, fungi, or parasites. The timeline of these discoveries is highlighted in Fig. 6 [12, 30, 59,60,61,62,63,64,65,66,67,68].

Timetable illustrating important discoveries in MCET biology. Four years after the first description of NETs by Brinkmann et al., MCETs were identified by Köckritz-Blickwede and colleagues in 2008. Over the past 5 years, a variety of stimuli for MCET formation have been described, including various microbes, microbial products, and chemicals, and several neoplastic MC cell lines and organ-derived MCs were reported to generate MCETs
General Function and Composition of MCETs
The formation of MCETs is mainly considered to constitute an extracellular mechanism of host defense against invading pathogens [66]. A variety of stimuli have been found to induce the formation of MCETs, including several cytokines such as IL-23 and IL-1β that not only stimulate the formation of MCETs but also trigger MC degranulation [69]. Both intracellular and extracellular microbes, their products, can induce the release of MCETs [59]. Additionally, chemicals including PMA and endogenous molecules, namely, H2O2 and glucose oxidase induce the production of MCETs [12]. In contrast to eosinophils and monocytes which form small ETs from mitochondrial DNA, MCs similar to neutrophils form ETs via release of nuclear DNA [8, 11, 62, 70]. First studies provided insights into the structure of MCETs as web-like DNA strands decorated with histones, tryptase, and the cathelicidin LL-37 [12]. Cathelicidins serve as a group of peptides with antimicrobial properties and are produced by several immune cells, epithelial and genital cells [71]. LL-37 is a cationic peptide produced in humans from its precursor molecule hCAP18 by kallikreins [72]. LL-37 is produced by epithelial cells of various tissues [72, 73], and enhances the function of neutrophils, induces the production of inflammatory chemokines including IL-8, and induces tissue vascularization [74] by stimulating angiogenesis [75]. LL-37 can activate and degranulate LAD2 cells, a mast cell line, and hematopoietic CD34 + derived MCs expressing MrgX2, the receptor for LL-37 [76]. Immunohistochemical studies show that exogenous LL-37 is taken up by LAD2 cells and can be detected in their cytoplasm and nuclei. Treatment of LAD2 cells with LL-37 induces the release of nucleic acids and at high doses reduces the viability of the treated cells [61]. Following its release, LL-37 induces the expression of a variety of TLRs in MCs including both surface-expressed TLRs such as TLR2, TLR4, or TLR5, and endosomal TLRs including TLR7 and TLR9 [72]. In addition, LL-37 upregulates retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) in peritoneal MCs [77]. Another element of MCETs is tryptase which exerts its effects mainly by proteolytic cleavage of protease-activated receptor (PAR)-2 which is expressed in various immune cells but also in endo- or epithelial cells [78]. In addition to signaling via PAR-2, tryptase has additional important biofunctions as a protease including activation of matrix metalloproteinases and degradation of extracellular matrices [79].
Imaging Techniques for the Study of MCETs
Prior to discussing the functional role of MCETs in microbial defense, we will briefly summarize the most relevant techniques used for the study of MCETs. Immunofluorescence and electron microscopy have been used successfully to demonstrate the ability of MCs to release ETs in the following protocol: (1) treatment of murine BMMCs (bone marrow–derived mast cells) or HMC-1 (Human Mast cell line-1) cells with PMA or glucose oxidase; (2) infection with S pyogenes that is either carboxyfluorescein labeled (if the imaging system is confocal microscopy) or unlabeled (if imaging is performed by electron microscopy); (3) fixation of cells using paraformaldehyde and washing in PBS for fluorescence microscopy; (4) applying antibodies against LL-37, tryptase, and histone; and (5) staining of DNA using DAPI (4,6-diamino-2-phenylindole) (or SYTOX-Green [59]) [12]. Alternatively, transmission electron microscopy has been used to study the structure and function of MCETs. To this end, samples are typically treated with MCET-inducing triggers such as PMA or bacteria and then fixed with glutaraldehyde-paraformaldehyde and osmium tetroxide. Following dehydration by ethanol samples are embedded in a mixture of ethanol-Epon and Epon resin. Additionally, uranyl acetate and lead citrate may be used to improve contrast [59]. Based on the finding that GreenGlo™ discriminates between nuclear DNA and strands of ETs by different excitation and emission wavelengths, Proust and colleagues developed a single-step protocol without washing to stain and discriminate these two types of DNA. Specifically in this protocol, nuclear DNA is detected by GreenGlo™ when excited at 470 nm with emission at 530 nm, while the same dye detects DNA strands of ETs at excitation of 350 nm and emission at 450 nm [80].
Role in Microbial Defense
Role in Anti-bacterial Defense
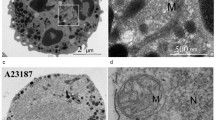
In their first report of MCETs, Köckritz-Blickwede and colleagues observed that S. pyogenes become entrapped by extracellular structures around MCs [12]. Subsequent studies revealed that MCETs are formed in response not only to extracellular bacteria but also to intracellular bacteria including L. monocytogenes. MC activation upon exposure to L. monocytogenes is mediated largely by listeriolysin with MCs releasing in response a cocktail of cytokines, mainly IL-1β, IL-6, IL-2, IL-4, IL-13, GM-CSF, and a variety of chemokines including CCL2, CCL3, CCL4, and CCL5. Additionally, the release of osteopontin from activated MCs contributes to the clearance of the bacteria [81]. In parallel, L. monocytogenes induces the formation of MCETs in HMC-1 cells as demonstrated by the release of nuclear DNA and examination of the nuclear envelope showed the separation of the inner and outer membranes (Fig. 7a). Enterococcus faecalis infection has gained growing relevance due to resistance of the bacteria to various antibiotics and as the cause of nosocomial infection with a mortality rate of above 50% in critically ill hospitalized patients. Scheb-Wetzel and colleagues assessed the production and activation of MCETs in E. faecalis infected primary bone marrow–derived murine MCs using a β-hexosaminidase assay and toluidine staining. The authors also investigated the release of IL-6 and TNF-α (the release of which is dependent upon TLR-2) and showed that exposure to E. faecalis activates MCs and induces the degranulation and the release of IL-6 and TNF-α [66]. Injection of GFP-expressing E. faecalis into mice and subsequent immunohistochemical staining for CD117 identified an interaction of E. faecalis and MCs in vivo. Experimental E. faecalis infection in MCs derived from TLR2−/− or MyD88−/− mice further revealed the importance of TLR2 and MyD88 in the effective response to E. faecalis by the release of antimicrobial peptides. Moreover, application of endonuclease resulted in destruction of MCETs (and a partial growth inhibitory effect of MCs on E. faecalis) [66]. In parallel, the low rate of internalized E. faecalis by MCs indicated the involvement of an extracellular mechanism in the elimination of the pathogen. Immunostaining for histones next revealed that MCs cocultured with E. faecalis produce MCETs and confocal microscopy demonstrated that ensnared E. faecalis were killed, presumably as a direct consequence of their entrapment in MCETs [66]. MCETs also seem to play a role in the elimination of S. aureus, as both bone marrow–derived murine mast cells (BMMC) and HMC-1 have been shown to release MCETs upon exposure to this pathogen. Interestingly, the bacterium seems to induce its own phagocytosis (partially through interaction between MCs expressing α5Β1 and fibronectin-binding proteins FnBPA and FnBPB [82]) in an attempt to evade being trapped and killed in MCETs [83, 84] (Fig. 7b).

Production of MCETs in response to intra/extracellular bacteria. a After being phagocytosed, L. monocytogenes become trapped in the phagosome. Listeriolysin becomes activated at the acidic pH of the phagosome and lyses it, allowing L. monocytogenes to escape into the cytosol. MCs in return release a wide spectrum of cytokines and chemokines and produce MCETs. Group A Streptococcus (GAS) stimulates the production of MCETs with LL-37 playing a crucial role in the structure and function of the extracellular traps. b S. aureus induces the production of MCETs; however, it uses a molecular mechanism to evade elimination by MCETs in that it induces its phagocytosis into the MC cytoplasm through interaction of FnBPA/FnBPB on S. aureus with fibronectin (as bridging molecule) and α5Β1 on MCs. Additionally, TLR2 and MyD88 play a role in recognition and signaling, respectively, when MCs are exposed to E. faecalis and produce MCETs in response
Role in Anti-fungal Defense
MCs can detect fungi like C. albicans via receptors such as β-glucan recognizing receptors (e.g., Dectin-1) and in response, release a variety of mediators including tryptase, histamine, prostaglandins (PGs), leukotrienes (LTs), and various cytokines, mainly CCL3, CCL4, TNF-α, IL-6, and IL-10 [65, 85] (Fig. 8a). The critical role of Dectin-1 in the recognition and response to C. albicans was highlighted in studies of cultured BMMCs from Dec−/− mice, which showed only an impaired release of TNF-α, IL-6, and IL-13 as compared to control BMMCs following stimulation with C. albicans yeast and hyphae [86]. Lopes and colleagues studied the mechanisms by which MCs limit the growth of C. albicans and reported that MCs produce MCETs. By measuring β-hexosaminidase, the authors showed that MCs become activated and degranulate when exposed to C. albicans. C. albicans-infected HMC-1 cells were shown to release not only IL-8 (acting as neutrophil chemoattractant), macrophage migration inhibitory factor (MIF), and IL-16 (acting as chemoattractant for CD4 + T lymphocytes), but also MCETs evident as DNA decorated with tryptase after 7 h [65]. To investigate the impact of MCETs the authors applied DNase prior to infection (Fig. 8b). However, C. albicans viability did not differ significantly in the presence vs. absence of DNase, suggesting that although MCs could ensnare C. albicans by MCETs, this mechanism may not play a major role in fungal elimination [65] (Fig. 8c).

Main mechanisms used by MCs to control C. albicans infection. a C. albicans are recognized by MCs upon engaging MC surface expressed Dectin-1, MCs in turn release mediators including tryptase, histamine, PGs, LTs, CCL3, CCL4, TNF-α, IL-6, and IL-10. b Upon recognizing C. albicans, MCs become activated and degranulate and release IL-8 (neutrophil chemoattractant), MIF, and IL-16 (chemoattractant for CD4 + T lymphocytes); c comparing C. albicans viability either in the presence or absence of DNase showed no significant difference suggesting that MCET formation is not the main extracellular mechanism of C. albicans elimination
Role in Anti-parasitic Defense
Formation of MCETs has also been reported to play a role in the defense against parasites. To this end, Naqvi and coworkers investigated the elimination of Leishmania donovani and Leishmania tropica by peritoneal MCs (PMCs) and Rat Basophilic Leukaemia (RBL-2H3) cells. The authors reported a significant decrease in the viability rate of RBL-2H3 cells cocultured with either L. tropica or L. donovani promastigotes [63]. To probe for the release of MCETs, RBL-2H3 cells were seeded on cover slides and then co-cultured with carboxyfluorescein N-succinimidyl ester (CFSE) labeled promastigotes of L. donovani and L. tropica for 24 h. DNA was stained by DAPI, and fluorescently tagged antibodies were used to determine the presence of tryptase and histones. Treatment with DNase increased the viability of promastigotes demonstrating the functional relevance of MCETs in the anti-parasitic defense [63]. The results of this study showed that formation of MCETs was an extracellular mechanism used by MCs to eliminate leishmaniosis infection. However, coculturing RBL-2H3 with L. donovani and L. tropica could decrease the viability of the cells when compared to the control group after 18 h; in which, for example, coculturing the with promastigotes of L. tropica showed a decrease in cell viability (89.5% ± 2.5%; at 18 h and 79.3% ± 3.5% at 24 h) when compared to the control group (96.2% ± 3%). This group of researchers, to confirm the death of MCs during the production and the release of MCETs, investigated the presence of extracellular DNA using Sytox Green staining after co-culturing MCs with the promastigotes of L. donovani and L. tropica. Their results showed that only 2.3% ± 1.5% of MCs cultured in the absence of parasites released extracellular DNA after 18 h, while 6.5% ± 0.5% of the cocultured MCs with L. tropica did so. Interestingly, the rate increased and 21.6% ± 1.2% of MCs were reported to release extracellular DNA only after 24 h [63].
Regulation of MCET Formation
As compared to the process of NETosis, the insight into the molecular mechanisms regulating the formation of MCETs is still sparse. In one of the few mechanistic studies, Möllerherm and colleagues recently demonstrated the formation of MCETs in response to short-term hypoxia (3 h). Notably, formation of MCETs in response to hypoxia was independent of hypoxia-inducible factor 1α (HIF-1α), a transcription factor that is critically involved in the adaptation to hypoxia. At normoxia, HIF-1α is rapidly degraded via the proteasome but stabilized when the cells experience hypoxia resulting in the transcription of hypoxia-regulated genes including erythropoietin, glucose transporters, glycolytic enzymes, antimicrobial factors, and VEGF [68]. While it has previously been reported that HIF-1α may induce the formation of MCETs [64], hypoxia caused MCET formation via a HIF-1α independent mechanism while suppressing the release of proinflammatory mediators including TNF-α, possibly in an attempt to attenuate the development of an inflammatory state and thus, to prevent tissue injury during hypoxia [64].
Unmet Questions: Themes for Further Investigations
In this section, we highlight major unmet questions in the structure, biology, function, and regulation of MCETs as important topics for further investigations in the field (Table 2).
Summary and Conclusion
Following the initial discovery of NETs in 2004, a similar ability for ETosis—albeit at a smaller scale—was demonstrated in various myeloid cells including eosinophils and monocytes by ejection of mitochondrial DNA. In contrast, mast cells seem to be the only other immune cell identified so far that is—similar to neutrophils—able to form ETs from nuclear DNA. Engagement of receptors by various ligands and also chemicals induces the formation of MCETs. The main inducers and involved receptors are listed in Table 3.
It should be noticed that the shape of MCETs seems to differ according to the local tissue and testing environment which should be considered in the interpretation of results. Specifically, MCETs in skin specimen are more compact when compared to those formed in vitro [69]. Protocols for the investigation of ETs are overall similar for different innate immune cell populations, and the function of ETs is largely determined by the bioactivity and biofunction of peptides decorating the ejected DNA strands. While our understanding of their physiological and pathogenic role is still rudimentary—as compared to the well-established role of NETs—MCETs have recently become implicated in host defense as well as various autoimmune, cardiovascular, or pulmonary disorders. Like NETs, MCETs act as scaffolds composed of nuclear DNA and peptides with antimicrobial activity that act as extracellular mechanism for trapping and killing of invading pathogens. Although the production of extracellular traps by immune cells has been predominantly linked to antimicrobial defense, some lines of evidence suggest a link to other pathologic conditions. For instance, MCs have been found to infiltrate and degranulate in skeletal muscles in autopsy samples of patients with amyotrophic lateral sclerosis (ALS) and are associated with NET producing neutrophils by recruiting them via the release of chymase that acts as neutrophil chemoattractant. Interestingly, the application of masitinib (a widely used tyrosine kinase inhibitor) could suppress the axonal pathology and secondary demyelination in ALS by suppressing MCs and interference with their role in neutrophils recruitment [87]. Analogously, MCs have been shown to infiltrate lung and vascular tissue in pulmonary hypertension and lung fibrosis [88]. Notably, pathological remodeling in these diseases could be attenuated or prevented not only by mast cell stabilizers or in mast cell deficient animals [89, 90], but also—at least in vitro—by DNase treatment [91, 92], suggesting a potential pathogenic contribution of MCETs. Interrupting the formation of MCETs may also act as a successful strategy of pathogens to evade the MC-mediated immune response. Along these lines, MCET formation can be detected following stimulation with heat-killed Mycobacterium tuberculosis, yet not in response to its viable counterparts, as catalase from Mycobacterium tuberculosis seems to prevent MCET formation by degrading hydrogen peroxide [60]. Other pathogens such as C. albicans may evade entrapment by MCETs by expressing DNase as a virulence factor [57]. At present, our understanding of MCETs, their formation, and structure, as well as their involvement in microbial defense and non-infectious pathologies, is only beginning to emerge. Although formation of ETs including MCETs is likely to be a late response but effective one against the presence of intruding microorganisms, however, the release of DNA into extracellular space may orchestrate the immune responses such as the production of anti-citrullinated protein antibodies in seropositive rheumatoid arthritis. Not surprisingly, since many AMPs attached to DNA web should be normally restricted in cytoplasmic granules, their release may have harmful effects such as degrading ECM or activating tissue-destructive mechanisms [93, 94]. Better insight into the function and regulation of MCETs, as well as the mechanisms by which pathogens tend to evade MCET-mediated elimination may provide not only important biological insights but pave the way for novel interventions in infectious, autoimmune, and other mast cell-related diseases.
Abbreviations
- BMMCs:
-
Bone marrow derived mast cells
- DAPI:
-
4,6-Diamino-2-phenylindole
- ET:
-
Extracellular trap
- GAS:
-
Group A Streptococcus
- H3Cit:
-
Citrullinated histone H3
- HIF-1α:
-
Hypoxia-inducible factor 1α
- HMC-1:
-
Human Mast cell line-1
- HMDM:
-
Human monocyte–derived macrophage
- mAb:
-
Monoclonal antibody
- MCETs:
-
MCs extracellular trap
- mDCs:
-
Myeloid dendritic cells
- MPO:
-
Myeloperoxidase
- NOD:
-
Nucleotide-binding oligomerization domain
- PAD:
-
Peptidyl arginase deiminase
- PAD:
-
Peptidyl arginine deiminase
- PMA:
-
Phorbol-12-myristate-13-acetate
- RIG-I:
-
Retinoic acid-inducible gene I
- ROS:
-
Reactive oxygen species
- RSV:
-
Respiratory syncytial virus
- NLRP3:
-
NOD-like receptor protein 3
- ASC:
-
Apoptosis-associated speck-like protein
References
Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A (2017) Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 6. https://doi.org/10.7554/eLife.24437
Vorobjeva N, Galkin I, Pletjushkina O, Golyshev S, Zinovkin R, Prikhodko A, Pinegin V, Kondratenko I, Pinegin B (1866) Chernyak B (2020) Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochim Biophys Acta 5:165664. https://doi.org/10.1016/j.bbadis.2020.165664
Hoffmann JH, Enk AH (2016) Neutrophil extracellular traps in dermatology: caught in the NET. J Dermatol Sci 84(1):3–10. https://doi.org/10.1016/j.jdermsci.2016.07.001
Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A (2015) Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (Baltimore, MD) 62(2):600–614. https://doi.org/10.1002/hep.27841
Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, Luo L, Mörgelin M, Regner S, Thorlacius H (2015) Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology 149(7):1920-1931.e1928. https://doi.org/10.1053/j.gastro.2015.08.026
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, Liu J, Xing Y, Chen X, Su S, Song E (2020) DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583(7814):133–138. https://doi.org/10.1038/s41586-020-2394-6
von Köckritz-Blickwede M, Nizet V (2009) Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (Berl) 87(8):775–783. https://doi.org/10.1007/s00109-009-0481-0
Pertiwi KR, de Boer OJ, Mackaaij C, Pabittei DR, de Winter RJ, Li X, van der Wal AC (2019) Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J Pathol 247(4):505–512. https://doi.org/10.1002/path.5212
Brinkmann V (2018) Neutrophil extracellular traps in the second decade. J Innate Immun 10(5–6):414–421. https://doi.org/10.1159/000489829
Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S, de Chaisemartin L (2017) Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102(3):775–781. https://doi.org/10.1189/jlb.3MA0916-411R
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU (2008) Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14(9):949–953. https://doi.org/10.1038/nm.1855
von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E (2008) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111(6):3070–3080. https://doi.org/10.1182/blood-2007-07-104018
Elieh-Ali-Komi D, Cao Y (2017) Role of mast cells in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Rev Allergy Immunol 52(3):436–445. https://doi.org/10.1007/s12016-016-8595-y
Elieh Ali Komi D, Ribatti D (2019) Mast cell-mediated mechanistic pathways in organ transplantation. Eur J Pharmacol 857:172458. https://doi.org/10.1016/j.ejphar.2019.172458
Komi DEA, Redegeld FA (2020) Role of mast cells in shaping the tumor microenvironment. Clin Rev Allergy Immunol 58(3):313–325. https://doi.org/10.1007/s12016-019-08753-w
Elieh Ali Komi D, Wöhrl S, Bielory L (2020) Mast cell biology at molecular level: a comprehensive review. Clin Rev Allergy Immunol 58(3):342–365. https://doi.org/10.1007/s12016-019-08769-2
Elieh Ali Komi D, Grauwet K (2018) Role of mast cells in regulation of t cell responses in experimental and clinical settings. Clin Rev Allergy Immunol 54(3):432–445. https://doi.org/10.1007/s12016-017-8646-z
Elieh Ali Komi D, Rambasek T, Bielory L (2018) Clinical implications of mast cell involvement in allergic conjunctivitis. Allergy 73(3):528–539. https://doi.org/10.1111/all.13334
Maurer M, Khan DA, Elieh Ali Komi D, Kaplan AP (2021) Biologics for the use in chronic spontaneous urticaria: when and which. J Allergy Clin Immunol Pract 9(3):1067–1078. https://doi.org/10.1016/j.jaip.2020.11.043
Kikuchi-Ueda T, Kamoshida G, Ubagai T, Nakano R, Nakano A, Akuta T, Hikosaka K, Tansho-Nagakawa S, Kikuchi H, Ono Y (2017) The TNF-alpha of mast cells induces pro-inflammatory responses during infection with Acinetobacter baumannii. Immunobiology 222(11):1025–1034. https://doi.org/10.1016/j.imbio.2017.05.015
Liu C, Ouyang W, Xia J, Sun X, Zhao L, Xu F (2018) Tumor necrosis factor-alpha is required for mast cell-mediated host immunity against cutaneous Staphylococcus aureus infection. J Infect Dis 218(1):64–74. https://doi.org/10.1093/infdis/jiy149
Komi DEA, Mortaz E, Amani S, Tiotiu A, Folkerts G, Adcock IM (2020) The role of mast cells in IgE-independent lung diseases. Clin Rev Allergy Immunol 58(3):377–387. https://doi.org/10.1007/s12016-020-08779-5
Nakano N, Nishiyama C, Yagita H, Koyanagi A, Akiba H, Chiba S, Ogawa H, Okumura K (2009) Notch signaling confers antigen-presenting cell functions on mast cells. J Allergy Clin Immunol 123(1):74-81.e71. https://doi.org/10.1016/j.jaci.2008.10.040
Suurmond J, Dorjee AL, Huizinga TW, Toes RE (2016) Human mast cells costimulate T cells through a CD28-independent interaction. Eur J Immunol 46(5):1132–1141. https://doi.org/10.1002/eji.201545914
Merluzzi S, Frossi B, Gri G, Parusso S, Tripodo C, Pucillo C (2010) Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA-secreting plasma cells. Blood 115(14):2810–2817. https://doi.org/10.1182/blood-2009-10-250126
Breitling S, Hui Z, Zabini D, Hu Y, Hoffmann J, Goldenberg NM, Tabuchi A, Buelow R, Dos Santos C, Kuebler WM (2017) The mast cell-B cell axis in lung vascular remodeling and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 312(5):L710-l721. https://doi.org/10.1152/ajplung.00311.2016
Pinke KH, Lima HG, Cunha FQ, Lara VS (2016) Mast cells phagocyte Candida albicans and produce nitric oxide by mechanisms involving TLR2 and Dectin-1. Immunobiology 221(2):220–227. https://doi.org/10.1016/j.imbio.2015.09.004
Alvendal C, Ehrström S, Brauner A, Lundberg JO, Bohm-Starke N (2017) Elevated nitric oxide in recurrent vulvovaginal candidiasis—association with clinical findings. Acta Obstet Gynecol Scand 96(3):295–301. https://doi.org/10.1111/aogs.13093
Min D, Shin MH (2009) NADPH oxidase-derived ROS mediates mast cell degranulation induced by secretory products secreted by Trichomonas vaginalis (133.5). 182 (1 Supplement):133.135–133.135
Pertiwi KR, De Boer OJ, Pabittei DR, Mackaaij C, De Winter RJ, Van Der Wal AC (2018) P373Macrophage, eosinophil, and mast cell extracellular traps (METs, EETs and MCETs) participate in coronary thrombus evolution after acute myocardial infarction. Cardiovasc Res 114(suppl_1):S95-S95. https://doi.org/10.1093/cvr/cvy060.283%JCardiovascularResearch
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17(3):151–164. https://doi.org/10.1038/nri.2016.147
Dhuriya YK, Sharma D (2018) Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation 15(1):199. https://doi.org/10.1186/s12974-018-1235-0
Frank D, Vince JE (2019) Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26(1):99–114. https://doi.org/10.1038/s41418-018-0212-6
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, Yu T, Wu X, Shi Y, Ma P, Shu Y (2020) Pyroptosis: a new frontier in cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 121:109595. https://doi.org/10.1016/j.biopha.2019.109595
Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF (2013) Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood 121(11):2074–2083. https://doi.org/10.1182/blood-2012-05-432088
Wu SY, Wu-Hsieh BA (2020) Neutrophil extracellular trap killing assay of Candida albicans. Bio-Protoc 10(16):e3716. https://doi.org/10.21769/BioProtoc.3716
White PC, Chicca IJ, Cooper PR, Milward MR, Chapple IL (2016) Neutrophil extracellular traps in periodontitis: a web of intrigue. J Dent Res 95(1):26–34. https://doi.org/10.1177/0022034515609097
Neeli I, Khan SN, Radic M (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol (Baltimore, Md : 1950) 180(3):1895–1902. https://doi.org/10.4049/jimmunol.180.3.1895
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184(2):205–213. https://doi.org/10.1083/jcb.200806072
Wartha F, Henriques-Normark B (2008) ETosis: a novel cell death pathway. Sci Signal 1(21):pe25. https://doi.org/10.1126/stke.121pe25
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science (New York, NY) 303(5663):1532–1535. https://doi.org/10.1126/science.1092385
Branitzki-Heinemann K, Mollerherm H, Vollger L, Husein DM, de Buhr N, Blodkamp S, Reuner F, Brogden G, Naim HY, von Kockritz-Blickwede M (2016) Formation of neutrophil extracellular traps under low oxygen level. Front Immunol 7:518. https://doi.org/10.3389/fimmu.2016.00518
Zhang F, Yang XM, Jia SY (2020) Characteristics of neutrophil extracellular traps in patients with periodontitis and gingivitis. Braz Oral Res 34:e015. https://doi.org/10.1590/1807-3107bor-2020.vol34.0015
Sharma R, O'Sullivan KM, Holdsworth SR, Bardin PG, King PT (2017) Visualizing macrophage extracellular traps using confocal microscopy. Journal of visualized experiments : JoVE (128). https://doi.org/10.3791/56459
Arelaki S, Arampatzioglou A, Kambas K, Sivridis E, Giatromanolaki A, Ritis K (2018) Mast cells co-expressing CD68 and inorganic polyphosphate are linked with colorectal cancer. PLoS One 13(3):e0193089. https://doi.org/10.1371/journal.pone.0193089
Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q, Tian J, Zhao H, Liu Z, Ma S, Zhang S, Novakovic VA, Shi J, Hu S (2020) Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine 53:102671. https://doi.org/10.1016/j.ebiom.2020.102671
Ueki S, Tokunaga T, Melo RCN, Saito H, Honda K, Fukuchi M, Konno Y, Takeda M, Yamamoto Y, Hirokawa M, Fujieda S, Spencer LA, Weller PF (2018) Charcot-Leyden crystal formation is closely associated with eosinophil extracellular trap cell death. Blood 132(20):2183–2187. https://doi.org/10.1182/blood-2018-04-842260
Silveira JS, Antunes GL, Gassen RB, Breda RV, Stein RT, Pitrez PM, da Cunha AA (2019) Respiratory syncytial virus increases eosinophil extracellular traps in a murine model of asthma. Asia Pac Allergy 9(4):e32. https://doi.org/10.5415/apallergy.2019.9.e32
Dworski R, Simon HU, Hoskins A, Yousefi S (2011) Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol 127(5):1260–1266. https://doi.org/10.1016/j.jaci.2010.12.1103
Choi Y, Le Pham D, Lee DH, Lee SH, Kim SH, Park HS (2018) Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp Mol Med 50(8):104. https://doi.org/10.1038/s12276-018-0136-8
Nyenhuis SM, Alumkal P, Du J, Maybruck BT, Vinicky M, Ackerman SJ (2019) Charcot-Leyden crystal protein/galectin-10 is a surrogate biomarker of eosinophilic airway inflammation in asthma. Biomark Med 13(9):715–724. https://doi.org/10.2217/bmm-2018-0280
Schulz M, Zambrano F, Schuppe HC, Wagenlehner F, Taubert A, Gaertner U, Sanchez R, Hermosilla C (2019) Monocyte-derived extracellular trap (MET) formation induces aggregation and affects motility of human spermatozoa in vitro. Systems biology in reproductive medicine 65(5):357–366. https://doi.org/10.1080/19396368.2019.1624873
Munoz-Caro T, Silva LM, Ritter C, Taubert A, Hermosilla C (2014) Besnoitia besnoiti tachyzoites induce monocyte extracellular trap formation. Parasitol Res 113(11):4189–4197. https://doi.org/10.1007/s00436-014-4094-3
Reichel M, Muñoz-Caro T, Sanchez Contreras G, Rubio García A, Magdowski G, Gärtner U, Taubert A, Hermosilla C (2015) Harbour seal (Phoca vitulina) PMN and monocytes release extracellular traps to capture the apicomplexan parasite Toxoplasma gondii. Dev Comp Immunol 50(2):106–115. https://doi.org/10.1016/j.dci.2015.02.002
Smirnova TG, Savochkina AY, Dolgushin II, Nikushkina KV, Samuseva IV (2018) Changes in functional activity of neutrophils and monocytes isolated from the peripheral blood of women at different phases of the menstrual cycle. Bull Exp Biol Med 166(2):222–224. https://doi.org/10.1007/s10517-018-4318-0
Aulik NA, Hellenbrand KM, Czuprynski CJ (2012) Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infect Immun 80(5):1923–1933. https://doi.org/10.1128/iai.06120-11
Loureiro A, Pais C, Sampaio P (2019) Relevance of macrophage extracellular traps in C. albicans killing. Front Immunol 10:2767. https://doi.org/10.3389/fimmu.2019.02767
Liu P, Wu X, Liao C, Liu X, Du J, Shi H, Wang X, Bai X, Peng P, Yu L, Wang F, Zhao Y, Liu M (2014) Escherichia coli and Candida albicans induced macrophage extracellular trap-like structures with limited microbicidal activity. PLoS One 9(2):e90042. https://doi.org/10.1371/journal.pone.0090042
Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Gonzalez-Jimenez M, Paredes-Vivas Y, Cerbulo-Vazquez A, Serafin-Lopez J, Garcia-Perez B, Ullrich SE, Flores-Romo L, Perez-Tapia SM, Estrada-Parra S, Estrada-Garcia I, Chacon-Salinas R (2017) Listeria monocytogenes induces mast cell extracellular traps. Immunobiology 222(2):432–439. https://doi.org/10.1016/j.imbio.2016.08.006
Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Rodriguez-Lopez GM, Soria-Castro R, Garcia-Perez BE, Puebla-Osorio N, Ullrich SE, Luna-Herrera J, Flores-Romo L, Sumano-Lopez H, Perez-Tapia SM, Estrada-Parra S, Estrada-Garcia I, Chacon-Salinas R (2018) Mycobacterium tuberculosis catalase inhibits the formation of mast cell extracellular traps. Front Immunol 9:1161. https://doi.org/10.3389/fimmu.2018.01161
Dahl S, Anders E, Gidlöf O, Svensson D, Nilsson BO (2018) The host defense peptide LL-37 triggers release of nucleic acids from human mast cells. Peptides 109:39–45. https://doi.org/10.1016/j.peptides.2018.10.001
Clark M, Kim J, Etesami N, Shimamoto J, Whalen RV, Martin G, Okumura CYM (2018) Group A Streptococcus prevents mast cell degranulation to promote extracellular trap formation. Front Immunol 9:327. https://doi.org/10.3389/fimmu.2018.00327
Naqvi N, Ahuja K, Selvapandiyan A, Dey R, Nakhasi H, Puri N (2017) Role of Mast Cells in clearance of Leishmania through extracellular trap formation. Sci Rep 7(1):13240. https://doi.org/10.1038/s41598-017-12753-1
Mollerherm H, Branitzki-Heinemann K, Brogden G, Elamin AA, Oehlmann W, Fuhrmann H, Singh M, Naim HY, von Kockritz-Blickwede M (2017) Hypoxia modulates the response of mast cells to Staphylococcus aureus infection. Front Immunol 8:541. https://doi.org/10.3389/fimmu.2017.00541
Lopes JP, Stylianou M, Nilsson G, Urban CF (2015) Opportunistic pathogen Candida albicans elicits a temporal response in primary human mast cells. Sci Rep 5:12287. https://doi.org/10.1038/srep12287
Scheb-Wetzel M, Rohde M, Bravo A, Goldmann O (2014) New insights into the antimicrobial effect of mast cells against Enterococcus faecalis. Infect Immun 82(11):4496–4507. https://doi.org/10.1128/iai.02114-14
Hayes SM, Biggs TC, Goldie SP, Harries PG, Walls AF, Allan RN, Pender SLF, Salib RJ (2020) Staphylococcus aureus internalization in mast cells in nasal polyps: characterization of interactions and potential mechanisms. J Allergy Clin Immunol 145(1):147–159. https://doi.org/10.1016/j.jaci.2019.06.013
Branitzki-Heinemann K, Okumura CY, Vollger L, Kawakami Y, Kawakami T, Naim HY, Nizet V, Von Kockritz-Blickwede M (2012) A novel role for the transcription factor HIF-1alpha in the formation of mast cell extracellular traps. Biochem J 446(1):159–163. https://doi.org/10.1042/bj20120658
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (Baltimore, Md : 1950) 187 (1):490–500. https://doi.org/10.4049/jimmunol.1100123
Kerstan A, Simon HU, Yousefi S, Leverkus M (2012) Extensive accumulation of eosinophil extracellular traps in bullous delayed-pressure urticaria: a pathophysiological link? Br J Dermatol 166(5):1151–1152. https://doi.org/10.1111/j.1365-2133.2012.10848.x
Bei Y, Pan LL, Zhou Q, Zhao C, Xie Y, Wu C, Meng X, Gu H, Xu J, Zhou L, Sluijter JPG, Das S, Agerberth B, Sun J, Xiao J (2019) Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med 17(1):42. https://doi.org/10.1186/s12916-019-1268-y
Agier J, Brzezinska-Blaszczyk E, Zelechowska P, Wiktorska M, Pietrzak J, Rozalska S (2018) Cathelicidin LL-37 Affects Surface and Intracellular Toll-Like Receptor Expression in Tissue Mast Cells. J Immunol Res 2018:7357162. https://doi.org/10.1155/2018/7357162
Kusaka S, Nishida A, Takahashi K, Bamba S, Yasui H, Kawahara M, Inatomi O, Sugimoto M, Andoh A (2018) Expression of human cathelicidin peptide LL-37 in inflammatory bowel disease. Clin Exp Immunol 191(1):96–106. https://doi.org/10.1111/cei.13047
Martin Jensen M, Jia W, Schults AJ, Ye X, Prestwich GD, Oottamasathien S (2018) IL-33 mast cell axis is central in LL-37 induced bladder inflammation and pain in a murine interstitial cystitis model. Cytokine 110:420–427. https://doi.org/10.1016/j.cyto.2018.05.012
Salvado MD, Di Gennaro A, Lindbom L, Agerberth B, Haeggström JZ (2013) Cathelicidin LL-37 induces angiogenesis via PGE2-EP3 signaling in endothelial cells, in vivo inhibition by aspirin. Arterioscler Thromb Vasc Biol 33(8):1965–1972. https://doi.org/10.1161/atvbaha.113.301851
Subramanian H, Gupta K, Guo Q, Price R, Ali H (2011) Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: resistance to receptor phosphorylation, desensitization, and internalization. J Biol Chem 286(52):44739–44749. https://doi.org/10.1074/jbc.M111.277152
Agier J, Rozalska S, Wiktorska M, Zelechowska P, Pastwinska J, Brzezinska-Blaszczyk E (2018) The RLR/NLR expression and pro-inflammatory activity of tissue mast cells are regulated by cathelicidin LL-37 and defensin hBD-2. Sci Rep 8(1):11750. https://doi.org/10.1038/s41598-018-30289-w
Bagher M, Larsson-Callerfelt AK, Rosmark O, Hallgren O, Bjermer L, Westergren-Thorsson G (2018) Mast cells and mast cell tryptase enhance migration of human lung fibroblasts through protease-activated receptor 2. Cell Commun Signal 16(1):59. https://doi.org/10.1186/s12964-018-0269-3
Tan H, Chen Z, Chen F, Yao Y, Lai Y, Xu W, Liu X (2018) Tryptase promotes the profibrotic phenotype transfer of atrial fibroblasts by PAR2 and PPARgamma pathway. Arch Med Res 49(8):568–575. https://doi.org/10.1016/j.arcmed.2018.12.002
Proust A, Levesque JC, Barat C, Sato S, Tremblay MJ (2018) A new tool for detection of extracellular traps. Methods and applications in fluorescence 6(3):037002. https://doi.org/10.1088/2050-6120/aac51b
Jobbings CE, Sandig H, Whittingham-Dowd JK, Roberts IS, Bulfone-Paus S (2013) Listeria monocytogenes alters mast cell phenotype, mediator and osteopontin secretion in a listeriolysin-dependent manner. PLoS One 8(2):e57102. https://doi.org/10.1371/journal.pone.0057102
Bingham RJ, Rudino-Pinera E, Meenan NA, Schwarz-Linek U, Turkenburg JP, Hook M, Garman EF, Potts JR (2008) Crystal structures of fibronectin-binding sites from Staphylococcus aureus FnBPA in complex with fibronectin domains. Proc Natl Acad Sci USA 105(34):12254–12258. https://doi.org/10.1073/pnas.0803556105
Abel J, Goldmann O, Ziegler C, Holtje C, Smeltzer MS, Cheung AL, Bruhn D, Rohde M, Medina E (2011) Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J Innate Immun 3(5):495–507. https://doi.org/10.1159/000327714
Agerer F, Michel A, Ohlsen K, Hauck CR (2003) Integrin-mediated invasion of Staphylococcus aureus into human cells requires Src family protein-tyrosine kinases. J Biol Chem 278(43):42524–42531. https://doi.org/10.1074/jbc.M302096200
Nieto-Patlan A, Campillo-Navarro M, Rodriguez-Cortes O, Munoz-Cruz S, Wong-Baeza I, Estrada-Parra S, Estrada-Garcia I, Serafin-Lopez J, Chacon-Salinas R (2015) Recognition of Candida albicans by Dectin-1 induces mast cell activation. Immunobiology 220(9):1093–1100. https://doi.org/10.1016/j.imbio.2015.05.005
De Zuani M, Paolicelli G, Zelante T, Renga G, Romani L, Arzese A, Pucillo CEM, Frossi B (2018) Mast cells respond to Candida albicans infections and modulate macrophages phagocytosis of the fungus. Front Immunol 9:2829. https://doi.org/10.3389/fimmu.2018.02829
Trias E, King PH, Si Y, Kwon Y, Varela V, Ibarburu S, Kovacs M, Moura IC, Beckman JS, Hermine O, Barbeito L (2018) Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 3(19). https://doi.org/10.1172/jci.insight.123249
Kuebler WM, Bonnet S, Tabuchi A (2018) Inflammation and autoimmunity in pulmonary hypertension: is there a role for endothelial adhesion molecules? (2017 Grover Conference Series). Pulmonary circulation 8(2):2045893218757596. https://doi.org/10.1177/2045893218757596
Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, Fujii H, Leong-Poi H, Kuppe H, Schermuly RT, Kuebler WM (2011) Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 37(6):1400–1410. https://doi.org/10.1183/09031936.00043310
Dahal BK, Kosanovic D, Kaulen C, Cornitescu T, Savai R, Hoffmann J, Reiss I, Ghofrani HA, Weissmann N, Kuebler WM, Seeger W, Grimminger F, Schermuly RT (2011) Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir Res 12(1):60. https://doi.org/10.1186/1465-9921-12-60
Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, Ainscough AJ, Hadinnapola C, Wilkins MR, Toshner M, Wojciak-Stothard B (2016) Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol 36(10):2078–2087. https://doi.org/10.1161/atvbaha.116.307634
Zhang S, Jia X, Zhang Q, Zhang L, Yang J, Hu C, Shi J, Jiang X, Lu J, Shen H (2020) Neutrophil extracellular traps activate lung fibroblast to induce polymyositis-related interstitial lung diseases via TLR9-miR-7-Smad2 pathway. J Cell Mol Med 24(2):1658–1669. https://doi.org/10.1111/jcmm.14858
Cooper PR, Palmer LJ (2000) Chapple IL (2013) Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol 63(1):165–197. https://doi.org/10.1111/prd.12025
Akiyama M, Zeisbrich M, Ibrahim N, Ohtsuki S, Berry GJ, Hwang PH, Goronzy JJ, Weyand CM (2019) Neutrophil extracellular traps induce tissue-invasive monocytes in granulomatosis with polyangiitis. Front Immunol 10:2617. https://doi.org/10.3389/fimmu.2019.02617
Huang H, Zhang H, Onuma AE, Tsung A (2020) Neutrophil elastase and neutrophil extracellular traps in the tumor microenvironment. Adv Exp Med Biol 1263:13–23. https://doi.org/10.1007/978-3-030-44518-8_2
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191(3):677–691. https://doi.org/10.1083/jcb.201006052
Martinod K, Witsch T, Farley K, Gallant M, Remold-O’Donnell E, Wagner DD (2016) Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. Journal of thrombosis and haemostasis : JTH 14(3):551–558. https://doi.org/10.1111/jth.13239
Guerra M, Halls VS, Schatterny J, Hagner M, Mall MA, Schultz C (2020) Protease FRET reporters targeting neutrophil extracellular traps. J Am Chem Soc. https://doi.org/10.1021/jacs.0c08130
O’Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE, Craik CS, Hermiston TW (2013) Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS One 8(9):e75141. https://doi.org/10.1371/journal.pone.0075141
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, Uehata T, Iwasaki H, Omori H, Yamaoka S, Yamamoto N, Akira S (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12(1):109–116. https://doi.org/10.1016/j.chom.2012.05.015
Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW, Libby P (2018) Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler Thromb Vasc Biol 38(8):1901–1912. https://doi.org/10.1161/atvbaha.118.311150
Gardiner EE, Andrews RK (2012) Neutrophil extracellular traps (NETs) and infection-related vascular dysfunction. Blood Rev 26(6):255–259. https://doi.org/10.1016/j.blre.2012.09.001
Elaskalani O, Abdol Razak NB, Metharom P (2018) Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun Signal 16(1):24. https://doi.org/10.1186/s12964-018-0235-0
Korkmaz B, Jenne DE, Gauthier F (2013) Relevance of the mouse model as a therapeutic approach for neutrophil proteinase 3-associated human diseases. Int Immunopharmacol 17(4):1198–1205. https://doi.org/10.1016/j.intimp.2013.07.003
Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, Pham CTN (2016) Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler Thromb Vasc Biol 36(8):1660–1669. https://doi.org/10.1161/atvbaha.116.307786
Kühnle A, Lütteke T, Bornhöfft KF, Galuska SP (2019) Polysialic acid modulates the binding of external lactoferrin in neutrophil extracellular traps. Biology 8(2). https://doi.org/10.3390/biology8020020
Okubo K, Kamiya M, Urano Y, Nishi H, Herter JM, Mayadas T, Hirohama D, Suzuki K, Kawakami H, Tanaka M, Kurosawa M, Kagaya S, Hishikawa K, Nangaku M, Fujita T, Hayashi M, Hirahashi J (2016) Lactoferrin suppresses neutrophil extracellular traps release in inflammation. EBioMedicine 10:204–215. https://doi.org/10.1016/j.ebiom.2016.07.012
Khadijeh N, Daniel Elieh Ali K, Habibolah K, Ali M, Asad V-R, Hamid Reza A, Mohammad Rasoul G, Amir K (2018) Investigation of serum levels and activity of matrix metalloproteinases 2 and 9 (MMP2, 9) in Opioid and Methamphetamine-Dependent Patients. Acta Med Iran 56(9)
Mirabdaly S, Elieh Ali Komi D, Shakiba Y, Moini A, Kiani A (2020) Effects of temozolomide on U87MG glioblastoma cell expression of CXCR4, MMP2, MMP9, VEGF, anti-proliferatory cytotoxic and apoptotic properties. Mol Biol Rep 47(2):1187–1197. https://doi.org/10.1007/s11033-019-05219-2
Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ (2015) Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis 74(7):1417–1424. https://doi.org/10.1136/annrheumdis-2013-204837
Shan Q, Dwyer M, Rahman S, Gadjeva M (2014) Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity. Infect Immun 82(10):4135–4143. https://doi.org/10.1128/iai.02169-14
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5(10):e1000639. https://doi.org/10.1371/journal.ppat.1000639
Fabisiak A, Murawska N, Fichna J (2016) LL-37: Cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacological reports : PR 68(4):802–808. https://doi.org/10.1016/j.pharep.2016.03.015
Neumann A, Völlger L, Berends ET, Molhoek EM, Stapels DA, Midon M, Friães A, Pingoud A, Rooijakkers SH, Gallo RL, Mörgelin M, Nizet V, Naim HY, von Köckritz-Blickwede M (2014) Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. J Innate Immun 6(6):860–868. https://doi.org/10.1159/000363699
Cao Y, Chen F, Sun Y, Hong H, Wen Y, Lai Y, Xu Z, Luo X, Chen Y, Shi J, Li H (2019) LL-37 promotes neutrophil extracellular trap formation in chronic rhinosinusitis with nasal polyps. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 49(7):990–999. https://doi.org/10.1111/cea.13408
Ávila EE, Salaiza N, Pulido J, Rodríguez MC, Díaz-Godínez C, Laclette JP, Becker I, Carrero JC (2016) Entamoeba histolytica trophozoites and lipopeptidophosphoglycan trigger human neutrophil extracellular traps. PLoS One 11(7):e0158979. https://doi.org/10.1371/journal.pone.0158979
Konstantinidis T, Kambas K, Mitsios A, Panopoulou M, Tsironidou V, Dellaporta E, Kouklakis G, Arampatzioglou A, Angelidou I, Mitroulis I, Skendros P, Ritis K (2016) Immunomodulatory role of clarithromycin in acinetobacter baumannii infection via formation of neutrophil extracellular traps. Antimicrob Agents Chemother 60(2):1040–1048. https://doi.org/10.1128/aac.02063-15
Jaeger SU, Schroeder BO, Meyer-Hoffert U, Courth L, Fehr SN, Gersemann M, Stange EF, Wehkamp J (2013) Cell-mediated reduction of human β-defensin 1: a major role for mucosal thioredoxin. Mucosal Immunol 6(6):1179–1190. https://doi.org/10.1038/mi.2013.17
Schroeder BO, Wu Z, Nuding S, Groscurth S, Marcinowski M, Beisner J, Buchner J, Schaller M, Stange EF, Wehkamp J (2011) Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature 469(7330):419–423. https://doi.org/10.1038/nature09674
Hu SC, Yu HS, Yen FL, Lin CL, Chen GS, Lan CC (2016) Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci Rep 6:31119. https://doi.org/10.1038/srep31119
Pietrocola G, Nobile G, Alfeo MJ, Foster TJ, Geoghegan JA, De Filippis V, Speziale P (2019) Fibronectin-binding protein B (FnBPB) from Staphylococcus aureus protects against the antimicrobial activity of histones. J Biol Chem 294(10):3588–3602. https://doi.org/10.1074/jbc.RA118.005707
Atiakshin D, Buchwalow I, Samoilova V, Tiemann M (2018) Tryptase as a polyfunctional component of mast cells. Histochem Cell Biol 149(5):461–477. https://doi.org/10.1007/s00418-018-1659-8
Murray DB, McLarty-Williams J, Nagalla KT, Janicki JS (2012) Tryptase activates isolated adult cardiac fibroblasts via protease activated receptor-2 (PAR-2). Journal of cell communication and signaling 6(1):45–51. https://doi.org/10.1007/s12079-011-0146-y
Church MK, Kolkhir P, Metz M, Maurer M (2018) The role and relevance of mast cells in urticaria. Immunol Rev 282(1):232–247. https://doi.org/10.1111/imr.12632
Anderson E, Stavenhagen K, Kolarich D, Sommerhoff CP, Maurer M, Metz M (2018) Human mast cell tryptase is a potential treatment for snakebite envenoming across multiple snake species. Front Immunol 9:1532. https://doi.org/10.3389/fimmu.2018.01532
Wang G, Narayana JL, Mishra B, Zhang Y, Wang F, Wang C, Zarena D, Lushnikova T, Wang X (2019) Design of antimicrobial peptides: progress made with human cathelicidin LL-37. Adv Exp Med Biol 1117:215–240. https://doi.org/10.1007/978-981-13-3588-4_12
Neumann A, Berends ET, Nerlich A, Molhoek EM, Gallo RL, Meerloo T, Nizet V, Naim HY, von Kockritz-Blickwede M (2014) The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem J 464(1):3–11. https://doi.org/10.1042/bj20140778
Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y, Men K, Qi X, Yi T, Wei Y, Wei X (2019) Induction of neutrophil extracellular traps during tissue injury: Involvement of STING and Toll-like receptor 9 pathways. Cell Prolif 52(3):e12579. https://doi.org/10.1111/cpr.12579
Afrin LB, Weinstock LB, Molderings GJ (2020) Covid-19 hyperinflammation and post-Covid-19 illness may be rooted in mast cell activation syndrome. International journal of infectious diseases : IJID: official publication of the International Society for Infectious Diseases 100:327–332. https://doi.org/10.1016/j.ijid.2020.09.016
Conti P, Caraffa A, Tetè G, Gallenga CE, Ross R, Kritas SK, Frydas I, Younes A, Di Emidio P, Ronconi G (2020) Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J Biol Regul Homeost Agents 34(5):1629–1632. https://doi.org/10.23812/20-2edit
Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, Saraiva EM, Porto BN (2018) Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep 8(1):14166. https://doi.org/10.1038/s41598-018-32576-y
Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, Kraft F, Lei Y, Fukasawa Y, Moeckel GW, Angelotti ML, Liapis H, Anders HJ (2017) Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol 28(6):1753–1768. https://doi.org/10.1681/asn.2016080925
Ehrens A, Lenz B, Neumann AL, Giarrizzo S, Reichwald JJ, Frohberger SJ, Stamminger W, Buerfent BC, Fercoq F, Martin C, Kulke D, Hoerauf A, Hübner MP (2021) Microfilariae trigger eosinophil extracellular DNA traps in a Dectin-1-dependent manner. Cell Rep 34(2):108621. https://doi.org/10.1016/j.celrep.2020.108621
Lin A, Rubin C, Khandpur R, Wang J, Riblett M, Yalavarthi S, Villanueva E, Shah P, Kaplan M, Bruce A (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (Baltimore, Md : 1950) 187:490–500. https://doi.org/10.4049/jimmunol.1100123
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V (2014) Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15(11):1017–1025. https://doi.org/10.1038/ni.2987
Elieh Ali Komi D, Sharma L, Dela Cruz CS (2018) Chitin and its effects on inflammatory and immune responses. Clin Rev Allergy Immunol 54(2):213–223. https://doi.org/10.1007/s12016-017-8600-0
Komi DE, Kazemi T, Bussink AP (2016) New insights into the relationship between Chitinase-3-Like-1 and asthma. Curr Allergy Asthma Rep 16(8):57. https://doi.org/10.1007/s11882-016-0637-2
Chumkaew P, Karalai C, Ponglimanont C, Chantrapromma K (2003) Antimycobacterial activity of phorbol esters from the fruits of Sapium indicum. J Nat Prod 66(4):540–543. https://doi.org/10.1021/np0204489
Bachiega TF, Dias-Melicio LA, Fernandes RK, de Almeida BH, Rodrigues DR, Ximenes VF, de Campos SÂM (2016) Participation of dectin-1 receptor on NETs release against Paracoccidioides brasiliensis: Role on extracellular killing. Immunobiology 221(2):228–235. https://doi.org/10.1016/j.imbio.2015.09.003
Muñoz-Caro T, Gibson AJ, Conejeros I, Werling D, Taubert A, Hermosilla C (2021) The role of TLR2 and TLR4 in recognition and uptake of the apicomplexan parasite eimeria bovis and their effects on NET formation. Pathogens (Basel, Switzerland) 10(2). https://doi.org/10.3390/pathogens10020118
Marcato LG, Ferlini AP, Bonfim RC, Ramos-Jorge ML, Ropert C, Afonso LF, Vieira LQ, Sobrinho AP (2008) The role of Toll-like receptors 2 and 4 on reactive oxygen species and nitric oxide production by macrophage cells stimulated with root canal pathogens. Oral Microbiol Immunol 23(5):353–359. https://doi.org/10.1111/j.1399-302X.2008.00432.x
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Daniel Elieh Ali Komi and Wolfgang M. Kuebler have been directly involved in the preparation of the manuscript. Daniel Elieh Ali Komi has designed and generated the figures. Wolfgang M. Kuebler has reviewed, revised, and added inputs.
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
No informed consent was required to prepare the manuscript.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elieh Ali Komi, D., Kuebler, W.M. Significance of Mast Cell Formed Extracellular Traps in Microbial Defense. Clinic Rev Allerg Immunol 62, 160–179 (2022). https://doi.org/10.1007/s12016-021-08861-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08861-6