
Abstract
Asthma is a heterogeneous and chronic inflammatory family of disorders of the airways with increasing prevalence that results in recurrent and reversible bronchial obstruction and expiratory airflow limitation. These diseases arise from the interaction between environmental and genetic factors, which collaborate to cause increased susceptibility and severity. Many asthma susceptibility genes are linked to the immune system or encode enzymes like metalloproteases (e.g., ADAM-33) or serine proteases. The S9 family of serine proteases (prolyl oligopeptidases) is capable to process peptide bonds adjacent to proline, a kind of cleavage-resistant peptide bonds present in many growth factors, chemokines or cytokines that are important for asthma. Curiously, two serine proteases within the S9 family encoded by genes located on chromosome 2 appear to have a role in asthma: CD26/dipeptidyl peptidase 4 (DPP4) and DPP10. The aim of this review is to summarize the current knowledge about CD26 and to provide a structured overview of the numerous functions and implications that this versatile enzyme could have in this disease, especially after the detection of some secondary effects (e.g., viral nasopharyngitis) in type II diabetes mellitus patients (a subset with a certain risk of developing obesity-related asthma) upon CD26 inhibitory therapy.
Similar content being viewed by others

Definition and Genetic Factors Implicated in Asthma
Asthma is a heterogeneous and chronic disease characterized by reversible expiratory airflow limitation, bronchial hyperresponsiveness, mucous cell hyperplasia, higher vasculature permeability, airway remodelling with fibrosis and inflammatory cell infiltration [1, 2]. Asthma is a major concern, with 334 million people worldwide affected, increasing prevalence [3] and a high economic charge [4]. It is more frequent and severe in boys until the age of 13 years, but both prevalence and severity of asthma rise in women after puberty, becoming even more prevalent in females [3, 5, 6]. Five to 10 % of cases display a highly severe and treatment-refractory disease, suffering from recurrent exacerbations that threaten a patient’s life and increase the healthcare costs. This is a complex pathology, with both genetic and environmental factors causing increased susceptibility and severity. Great efforts have been made to discover the genetic bases (mostly genome-wide association studies in asthma (GWAS)), revealing the influence of several genes encoding proteins with an important role in the immune system (HLA-DQ, HLA-G, IL1RL1, IL18R, TSLP, PDE, IL-33, LRRC32, SMAD3, IL2RB, IL6R, IL-13) and also proteases (ADAM33 and dipeptidyl peptidase 10 (DPP10)) [7–9].
Asthma and the Prolyl Oligopeptidase Family of Proteases: Dipeptidyl Peptidase 10 and CD26/DPP4
As commented above, proteases are involved in many physiological and pathological processes, including chronic respiratory conditions like asthma. Amongst all proteases, there are only a few proline-specific enzymes, as peptide bonds adjacent to proline (present in some growth factors, chemokines or cytokines) are resistant to cleavage [10]. These enzymes include serine proteases, with a serine residue in their catalytic region (sequence consensus Gly-Xaa-Ser-Xaa-Gly) [11] and covering different families (S1–S81) and subfamilies. Thus, the S9 family (prolyl oligopeptidases) includes from S9A to S9D, all with the catalytic triad Ser, Asp, His but with slightly different sequence consensus around the catalytic Ser. For example, most of the members of the S9B subfamily (EC 3.4.14.5; CD26/DPP4, DPP8, DPP9 and fibroblast activation protein/FAP/Seprase) (http://merops.sanger.ac.uk/) present the sequence Gly-Trp-Ser-Tyr-Gly-Gly, while the other additional two members, DPP6 (DPPX; Gly-Lys-Asp-Tyr-Gly-Gly) and DPP10 (Gly-Lys-Gly-Tyr-Gly-Gly), do not possess the catalytic serine and, therefore, DPP4 activity [12]. Curiously, four of these peptidases (CD26, FAP, DPP6, and DPP10) are type II membrane proteins released to the extracellular medium, three are encoded by genes located on chromosome 2 (CD26, FAP and DPP10) and only two (CD26, DPP10) appear to have a role in asthma [13–16]. For example, DPP10 has been linked to asthma susceptibility in different populations [7, 13, 15], and this protein (as well as DPP8 and DPP9) has been primarily located in the trachea and the bronchi of the airways in rats [17]. For its part, the protease CD26 is the major member of the S9B family and also a receptor for the Middle East respiratory syndrome coronavirus (MERS-CoV) [18, 19], a new coronavirus that causes severe lower respiratory tract infections that could lead to asthma exacerbations. CD26 has been found elevated in both plasma samples (soluble CD26 or sCD26) and the surface of peripheral CD4+ T cells from adult patients with allergic asthma [20, 21], and different animal models [14, 16, 22] suggest a role of this peptidase in the pathogenesis of this disease.
Structure and Distribution of CD26
Structure of the CD26 Molecule
CD26 is a single-pass type II integral membrane glycoprotein of 105–110 kDa [23]. Only the homodimeric form of CD26 is biologically active [24–27]. Homodimerization takes place in both the Golgi apparatus [28] and the endoplasmic reticulum [29]. Each monomer displays a highly conserved and short (six amino acids) cytoplasmic tail at the N-terminus, a 22-amino acid hydrophobic transmembrane region and a long extracellular domain of 738 amino acids. The heavily N-glycosylated extracellular domain can be subdivided, in turn, in several regions. The closest to the amino terminal part starts with a flexible stalk region and contains 8 out of 10 possible N-glycosylation sites, while the intermediate region is highly enriched in cysteines (9 out of 12). Finally, the active centre is located towards the carboxyl-terminal end and is another highly conserved region [30–34].
Tissue and Cell Distributions of CD26
Human CD26 is broadly distributed in a variety of cell types, tissues and organs [35–39]. Lungs display the second highest CD26 activity amongst organs, especially in lung parenchyma [17, 22]. Contrary to DPP8/9 and DPP10, bronchi almost do not express CD26 [17], but it can be found in the apical membrane of epithelial cells, capillary endothelial cells, fibroblasts and serosal submucosal glands of human bronchi [40–42]. In most of these places, CD26 levels are constitutive and highly correlated with the messenger RNA (mRNA) content (http://www.proteinatlas.org/ENSG00000197635-DPP4/tissue) [43]. However, IL-13 causes a strong proinflammatory upregulation of CD26 in the airway epithelial cells [44]. Moreover, both DPP4 activity and CD26 protein (but not mRNA) increase after allergen exposure in lung parenchyma in a rat model of allergic airway inflammation [17], suggesting that CD26 on lung epithelium could be important in asthma pathogenesis [45].
CD26 is also expressed in the immune system, being detected in medullar thymocytes, T cell areas of spleen and lymph nodes, peripheral blood T cells and, at a lesser extent, B cells, NK cells, monocytes/macrophages and granulocytes [24, 25, 30, 41, 46, 47]. CD26 density in these cells is heavily controlled and augments upon cell activation and acquisition of a memory phenotype, especially in the T cell lineage [33, 41, 48]. Human T lymphocytes display variable basal expressions of CD26 (CD4+ T >> CD8+ T cells), which depends on the individual and the mAb used. Despite that not all resting T cells are CD26+, most of them contain CD26 mRNA and display CD26 molecules on the surface 4–8 h after stimulation [49–51]. However, only a certain regulation on CD26 mRNA levels has been detected by either northern blot [36, 50, 52–54] or gene arrays [55] upon activation. Additionally, CD26 is upregulated by interferon gamma (IFNγ) on renal epithelial cells [56] and B-CLL [57] through the modification of mRNA levels.
Despite the above described adjustment of CD26 expression through mRNA levels, several evidences support the presence of upstream control levels. Thus, CD26 is strongly regulated at protein level by several soluble factors. For example, the TH1 cytokine IL-12 (and to a lesser extent IL-2) enhances the expression of CD26 on both activated T cells [58, 59] and NK cells (together with IL-15) [47, 60]. On the contrary, IFNγ (another TH1 cytokine) has no effect on CD26 levels in these lymphocytes [47, 58]. Regarding TH2 cytokines, IL-4 promotes the expression of CD26 on human B lymphocytes activated with Staphylococcus aureus Cowan I [61, 62], while this cytokine moves from a lack of effect [63] to a certain downmodulation of CD26 levels in T cells at high concentrations [unpublished results]. In addition, in vitro exposure to regulatory T cell (Treg)-derived soluble factors like transforming growth factor beta 1 (TGF-β1) [64, 65] or adenosine (Ado) [66] leads to diminished CD26 levels.
Most of peripheral blood Treg lymphocytes display an “activated-like” (e.g., CD25high), “memory” (CD45RO+) and an “anergic/apoptosis-prone” phenotype [67] and were expected to express other activation/memory markers such as CD26. This seems to be the case for rats, where CD25+ (Treg) and CD25− (effector T cells or Teff) subsets of peripheral CD4+ T cells show equivalent CD26 expression [68]. In contrast, human Treg cells (CD4+CD25high or CD4+FoxP3high) display lower levels of CD26 compared to Teff lymphocytes (CD4+CD25−/low or CD4+FoxP3−/low) [69–72]. This likely explains why cytokines that favour “effector” responses (e.g., IL-12) cause a strong upregulation of CD26 on “bulk” TH cell cultures, while others important for the Treg homeostasis (e.g., IL-2, IL-15) only induce a slight increase on CD26 levels [58, 73]. It also gives a clue on why CD26 is downmodulated upon exposure of T cells to TGF-β1 [64, 65].
CD26 expression amongst Teff lymphocytes is also variable, and TH17 cells appear to display the highest levels of CD26 according to the following order: TH17 >> TH1 > TH2 [72, 74–77]. Furthermore, two subsets were reported amongst TH17 cells on the base of CCR4 expression [78], but only the CCR4− TH17 subset (and not the TGF-β-secreting CCR4+ TH17 subpopulation) has a CD26high phenotype [79]. This finding likely indicates that the degree of phenotypic diversity found in Teff cells for CD26 levels is also probably present in Tregs and reflects the presence of functionally different subsets that mirror the corresponding Teff subsets [80].
The different expression levels of CD26 in Treg and Teff lymphocytes could depend on a variation in CD26 mRNA levels, like it happens with “regulatory-like” CD4+ T cells in classical Hodgkin’s lymphoma [81]. However, it is worth to mention that, apart from regulating this set of CD26 mRNA molecules, there is an intracellular pool of CD26 protein maintained by continuous translation in human T cells (regardless of their CD26+ or CD26− phenotype) [50] that can be mobilized towards the plasma membrane [54] or released into the extracellular space. Therefore, there could be a number of additional mechanisms leading to a CD26−/low phenotype in human Treg cells.
Body Fluid Compartments and Distribution of Soluble CD26
CD26 can be found in the extracellular space, with autocrine, paracrine or endocrine effects. The soluble version of CD26 (sCD26) has been described in many biological fluids (e.g., serum, plasma, synovial fluid, cerebrospinal fluid) from different organisms [82]. Bronchoalveolar lavage contains DPP4 enzymatic activity in rats [17] and humans [83]. In serum or plasma, at least 90 % of this activity is associated to a heavily glycosylated 110-kDa CD26 isoform [84, 85], whose concentration displays a normal distribution [86] and high biological variability [87, 88] that seems to depend on pre-analytical variables, like age or gender. Thus, serum sCD26 concentration increased up to 10–12 years in children, after which the values start decreasing [89]. In adults, a slight decrease of sCD26 or DPP4 activity in serum with age has been also detected [87, 88], but other studies did not describe such correlation [86, 90]. In addition, no differences were detected regarding gender in some studies [90], while different authors did find a higher sCD26 concentration in serum/plasma samples [86–89, 91] but a lower CD26 expression on CD4+ T cells (our unpublished results) from males.
Several studies have found a positive correlation between sDPP4 activity and sCD26 in humans [91–93], but others have reported a low correlation instead, with different explanations like the presence of hypersialylated CD26 isoforms or alternative proteins with DPP4 activity [82]. Thus, plasma samples contain a soluble glycoprotein named DPPT-L or attractin without homology with CD26 but with DPP4 activity [94–96], although this activity has been questioned more recently [97]. Attractin is accumulated in the plasma membrane upon T cell receptor (TCR) triggering (∼24 h) [95, 96], especially in CD26high Teff cells (our unpublished results), and released into the plasma at 48–72 h [95, 96]. Curiously, linkage disequilibrium studies show association of asthma with a region including the attractin (ATRN) gene, which is upstream of the gene encoding the secretase/sheddase ADAM-33 (an asthma susceptibility gene) [97].
Release of CD26 could be accomplished by a classical or non-classical pathway and either by a constitutive or induced mechanism. The classical pathway requires vesicular traffic from the endoplasmic reticulum/Golgi towards the plasma membrane. Intravesicular proteins are liberated in the extracellular medium after membranes merge (exocytosis), while transmembrane proteins appear in the secretome through proteolysis mediated by “secretases”/“sheddases”. This last process (“shedding”) delivers growth factors, cytokines, receptors and probably molecules like CD26 [98]. This is additionally supported by the lack of posttranscriptional splicing in CD26 mRNA, the absence of cytoplasmic and transmembrane domains in sCD26 or results from pulse chase and transfection experiments [25, 82]. However, CD26 has been detected in microvesicles and exosomes from lymphocytes (http://www.exocarta.org). Therefore, constitutive or induced release of CD26+ vesicles (exosomes, microvesicles or apoptotic bodies) into the medium could also contribute to the pool of sCD26 in the circulation and reflect the cell subset of origin (“cell lineage fingerprint”).
The cell source of sCD26 is still controversial [25, 82]. Visceral fat, at least in obese patients, overexpresses CD26 and releases this “adipokine” into the circulation [88], and there are data supporting the presence of a positive correlation between fasting sCD26 levels and body mass indexes (BMIs) >25 [88, 99]. Soluble CD26 may also originate from other sources, such as endothelial cells (e.g., lung) or epithelial cells from liver (bile canaliculi) or kidney. However, immune cells are also a likely source [25, 82, 100]. Thus, a transplantation model in rats has determined that, under healthy conditions, bone marrow-derived cells represent a significant source for sCD26 [101]. Therefore, the concentration of sCD26 could also be influenced by the number of lymphocytes [102] or mirror the predominant phenotype of circulating CD4+ T lymphocytes in a pathological situation.
CD26 and Asthma
Asthma Phenotypes, Disease Severity and CD26 levels in CD4+ T Cells
CD4+ TH cells play a central role in adaptive immune responses and the induction/persistence of asthma and other allergic/atopic diseases. They are plastic and heterogeneous lymphocytes, with four main lineages (TH1, TH2, TH17 and Treg cells) and different roles. For example, TH2 (GATA-3+) cells infiltrate airways in allergic asthma and produce a set of cytokines (IL-4, IL-5, IL-13) important for airway remodelling, leucocytosis, eosinophilia, macrophage/mast cell activation and B cell-dependent IgE elevation [103, 104]. In the same way that TH lymphocytes are heterogeneous, there are also a number of asthma phenotypes as a likely reflection of the TH cell heterogeneity itself, with both TH2high and TH2low phenotypes sharing different clinical signs: (a) the early allergic asthma, with a strong familiar background and mediated by TH2 cytokines (TH2high) and allergen-specific IgE (atopy); (b) the less-allergic late-onset asthma (TH2high), which is characterized by eosinophilic inflammation, absence of specific IgE (i.e., non-atopic) and worse response to corticosteroids; (c) the neutrophilic asthma, a TH2low phenotype refractory to corticosteroids and linked to TH17 responses, sputum neutrophilia and augmented levels of IL-8 and IL-17; (d) and the obesity-related asthma, a TH2low phenotype predominant in obese women, with higher levels of tumor necrosis factor alpha (TNFα), IL-6 and leptin but low numbers of eosinophils [1] (Table 1). Therefore, alternative CD26+/high effector TH subpopulations are gaining importance in asthma, particularly as disease becomes chronic and refractory to treatment. In this context, even though it was initially described that CD26+ TH1 cells were protective (“hygiene hypothesis”), now it is considered that these cells could be favouring the inflammation of the airways [105]. In addition, proinflammatory CD26high TH17 cells have been detected in biopsies from asthma patients and their cytokines (e.g., IL-17, IL-6, IL-21, IL-22) are involved in eosinophilia/neutrophilia and higher severity during acute attacks [106]. On the other hand, Treg cells (FoxP3+, CD26−/low) also seem to play a relevant role in controlling exaggerated TH2 responses and asthma development [9, 107, 108]. Treg cells exert their suppressor activity by using both soluble (e.g., adenosine, TGF-β, IL-10) and membrane-associated (e.g., TGF-β, CTLA-4) molecules [67], and a number of GWAS in asthma have identified some genes (e.g., IL2RB, SMAD3, GARP/LRRC32) linked to this suppressor activity. Thus, the lower or higher prevalence of different Teff subsets and/or the distortion of the Teff/Treg balance could also alter the phenotype and severity of asthma [104, 109, 110]. Moreover, depending on the asthma phenotype or the severity of this disease, the levels and functions of CD26 on the surface of immune cells and biofluids could be rather different and be a factor influencing disease development. Therefore, it is time to ask what the biological functions of CD26 are and what their connection with asthma is.
Enhancing Functions of Both Membrane and Soluble CD26 on Asthma
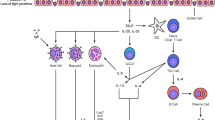
Functionally speaking, CD26 is a complex molecule with both harmful and beneficial activities in relation to asthma. With regard to the first ones, CD26 has been traditionally linked to both the amplification of TCR-mediated stimulatory signals and cell adhesion/migration. These functions could be dependent on production of certain biomolecules, the enzymatic activity of CD26 or the extracellular association with other proteins: adenosine deaminase (ADA) [111], CD45 [112], caveolin-1 [113], CXCR4 [114], collagen [115], plasminogen 2 [116], glypican-3 [117] or fibronectin III [118, 119] (Fig. 1). For example, CD26-mediated costimulation plays a role in the migration of mesothelioma cells through upregulated production of a current biomarker of “TH2high” asthma: periostin [120, 121]. CD26 location could also be important for this harmful function of CD26 in asthma. As a raft resident protein [112, 122], CD26 improves the immunological synapse formation between antigen-presenting cells (APCs) and T cells [112, 113], important for TH cell proliferation/differentiation and to empower these lymphocytes to proportionate help to B cells and produce immunoglobulins like IgE [30] (Fig. 1). This raft-dependent costimulatory function of CD26 could be mediated by other proteins like CD45, a tyrosine phosphatase (PTPase) essential for TCR signalling [123]. Cross-linking with anti-CD26 antibodies in T lymphocytes drives the recruitment to rafts and the interaction of both CD26 and CD45RO [112], which probably causes CD45 dimer dissociation [112], increased PTPase activity [124] and a signal transduction cascade that both overlaps with and boost the TCR pathway [112, 125–128] (Fig. 1).

Functions of both membrane and soluble CD26 on asthma. This figure summarizes both positive (i.e., those that favour asthma development or exacerbation) and negative (i.e., those that prevent asthma development or exacerbation) functions of CD26 on this disease
This potentially detrimental function of CD26 to the lungs of a person with asthma seems to involve the active centre [112, 129], but not necessarily the DPP4 activity. Thus, CD26 on TH cells recognizes caveolin-1 and induces the upregulation of the proinflammatory B7.2/CD86 molecule in APCs [113, 129]. In T cells, CD26-caveolin-1 interaction induces the coalescence of lipid rafts, recruitment of CARMA1 and activation/nuclear translocation of the proinflammatory and “rapid acting” transcription factor NF-kB [130] (Fig. 1). Therefore, the CD26+/high phenotype of Teff cells may favour allergen-induced inflammation in lungs [131], while the CD26−/low phenotype of Treg cells may contribute to keep low levels of CD86 on APCs and trigger anergy/apoptosis in allergen-specific naïve T cells. However, all these findings about the costimulatory role of CD26 need further clarification, as either a partial or complete genetic deletion of CD26 or the use of CD26 inhibitors does not impact this function [132, 133].
The costimulatory actions of CD26 also could depend on the interaction with another enzyme: ADA [111, 134–136]. Most of ADA molecules in lymphocytes are cytosolic, but a small proportion (10 %) are associated with either CD26 [111, 134, 135] or Ado receptors (ARs: A1AR and A2BAR) on the T cell membrane (ecto-ADA) [114, 135–138]. Binding of ADA to CD26 takes place in human (but not murine) lymphocytes [25, 63], and this interaction could potentially trigger a costimulatory signal that favours lymphocyte proliferation and TH1 cytokine and chemokine production [139–141] (Fig. 1). In turn, activation through the TCR/CD3 complex and costimulation with TH1 (but not TH2) cytokines increase the number of ecto-ADA molecules [63, 139] and the formation of the ADA-CXCR4-CD26 triad [114], which might help to trap the chemokine receptor CXCR4 at the immunological synapse to make lymphocytes insensitive to the chemokine stromal cell-derived factor α (SDF1α/CXCL12) and trigger a proliferative and cytokine-secreting response [142] (Fig. 1).
Costimulatory effects could also come from sCD26, which is capable of enhancing the T cell-mediated reaction against recall antigens (e.g., tetanus toxoid) or suboptimal amounts of polyclonal stimuli (PHA or anti-CD3) [143, 144]. Contrary to initial results [143], the most recent data point out that this proinflammatory effect of sCD26 is not dependent on CD26 or ADA-binding activities [145] and is likely triggered upon interaction with proteins like caveolin-1 [113, 129, 130] or insulin-like growth factor II/mannose-6-phosphate (IGF-II/M6P) receptor [146, 147] in APCs or protease-activated receptor 2 (PAR2) in smooth muscle cells [148]. sCD26 promotes CD86 upregulation in APCs [113, 129, 146] and the generation of reactive oxygen species (ROS) and toll-like receptors [149] (Fig. 1).
CD26 is the major member of the prolyl oligopeptidase family and cleaves X-proline or X-alanine dipeptides from the N-terminus of different polypeptides, even though other amino acids are accepted with lower efficiency: Pro > Ala > Hyp>> Ser > Gly > Val > Leu [25]. Apart from the primary sequence, the 3D structure, size and substrate ionization (optimum pH 7.5–8.5) significantly influence the catalytic efficiency of CD26 [10, 94, 100, 150, 151]. Peptides containing X-proline or X-alanine at the N-terminus are not easy to process by most of proteases [152], which means that CD26 has a key biological role. Indeed, DPP4 activity has been linked to the activation of T lymphocytes in response to extracellular stimuli. For example, Jurkat cells transfected with CD26 display greater activation than those expressing DPP4-deficient CD26 molecules [153]. DPP4 activity is also necessary in sCD26 molecules to boost the tetanus toxoid-dependent proliferation [146]. CD26 inhibitors yield similar results in vitro, with suppression of T cell proliferation and secretion of proinflammatory cytokines but enhanced release of TGF-β1 [154–157]. In vivo, the use of CD26 inhibitors in murine models of human diseases like rheumatoid arthritis [158], multiple sclerosis [159] or asthma (aerosolized simultaneously with the allergen) [45] also describes a proinflammatory role of CD26. Moreover, CD26 inhibitors also support a positive role of CD26 in NLRP3 inflammasome formation [160]. However, enzyme activity is required, but not in absolute terms, during the costimulatory function of CD26, since this biological activity is retained after deleting part of the hydrolase domain [132] (Fig. 1).
Inhibitory Functions of Both Membrane and Soluble CD26 on Asthma
Apart from its costimulatory and asthma-detrimental role, there is also evidence that CD26 exhibits other asthma-preventive activities. This protective function of CD26 is linked to the indirect or direct control of different soluble mediators like Ado, bradykinin or neuropeptides that are involved in bronchoconstriction, inflammation, chemoattraction or airway remodelling in asthma patients [161]. For instance, the nucleoside Ado is the substrate of ADA, an enzyme that catalyses its irreversible deamination to inosine [139, 140]. As already mentioned, ADA has been found anchored to CD26 (Fig. 1) in many cells, but two frequently regarded proinflammatory ARs (A1AR and A2BAR) also interact with this enzyme, which seems important for their efficient ligand-dependent signalling [111, 134–138]. ARs are novel targets for treatment of human asthma, and some antagonists and agonists have entered the clinical phase with a not totally clear efficacy [161, 162]. Indeed, there is an elevated expression of asthma-favouring and ADA-interacting receptors A1AR and A2BAR in bronchial tissue [161, 162], but probably reduced amounts of asthma-protective A2AAR [161]. In addition, there is an augmented Ado concentration in the extracellular compartment in asthma. This nucleoside plays a detrimental role in this disease by causing bronchoconstriction, airway inflammation (e.g., mast cell degranulation, airway accumulation of eosinophils), airway remodelling, edema or mucus production [161, 162]. These harmful effects also depend on the specific engaged type I purinergic receptor and the particular cells expressing the ARs in airways (e.g., APCs, lymphocytes, eosinophils, neutrophils, endothelial and mast, epithelial, endothelial and smooth muscle cells) [161, 162]. This role of Ado in asthma is affected by binding affinity, receptor density and local levels of this nucleoside, which are influenced by a delicate balance where different mechanisms are involved (e.g., extracellular/intracellular Ado metabolism) [161, 162]. Amongst these mechanisms, the Ado catabolism mediated by ADA molecules anchored to CD26 is the major regulated pathway that influences the local concentration and biological effects of Ado. In this respect, relevant TH2 cytokines in allergic asthma (IL-4, IL-13) reduce ADA activity in the lung [161] and ecto-ADA levels on TH lymphocytes [63]. These findings suggest that low levels of CD26/ecto-ADA on the TH2 subset in TH2high (allergic/atopic) asthma could favour the local accumulation of Ado, the activation of A1AR and the participation of proinflammatory ARs that require Ado concentrations over the physiological levels (e.g., the low-affinity A2BAR) in order to neutralize the off-signals provided by the engagement of high-affinity and asthma-protective A2AARs. Therefore, it is likely that AR-targeted therapy is more effective in TH2high asthma than in TH2low or severe asthma, wherein CD26/ecto-ADAhigh TH1 and TH17 cells are more frequent. Moreover, additional studies are needed to clarify what could be exactly the role of Treg cells in asthma, a lymphocyte subset with a CD26/ecto-ADAlow phenotype that should favour the high production of Ado [70]. On the other hand, CD45 has been involved in Janus kinase dephosphorylation [163]. Therefore, the CD26-CD45 association could have a negative and DPP4-independent role important to restrain the signal transduction of cytokine receptors [122] (Fig. 1). In this sense, the low PTPase activity in human naïve CD4+CD45RA+CD26−/low T cells makes them susceptible to small amounts of IL-4, as mentioned as an important cytokine in allergic asthma, while memory/effector CD4+CD45RO+CD26+/high T cells secrete this soluble factor upon activation, but are less sensitive to its effects [164]. Indeed, high levels of CD26 in memory/effector TH lymphocytes might act as a “brake” for the proliferative responses to cytokines (for example, IL-12; our unpublished results).
The protective function of CD26 in asthma could also be linked to the DPP4 activity (Fig. 1). Some CD26 substrates fall outside the scope of the immune system and this review, like incretins (glucose-dependent insulinotropic peptide (GIP), glucagon-like peptide-1 (GLP-1), glucagon-like peptide-2 (GLP-2)) [10, 165, 166], glucagon, pituitary adenylate cyclase-activating polypeptide (PACAP), gastrin-releasing peptide (GRP), peptide YY, vasoactive peptides (bradykinin and vasoactive intestinal peptide (VIP)), natriuretic peptides (B-type natriuretic peptide (BNP)) and neuropeptides (neuropeptide Y (NPY), beta-casomorphins, endomorphins, substance P) [41, 82, 166, 167] (Table 2). However, some others have an important immunomodulatory role, such as cytokines (e.g., IL-3, G-CSF, GM-CSF) [168] or chemokines (e.g., RANTES/CCL5, eotaxin/CCL11, MDC/CCL22) [82, 166]. CD26 enzymatic activity downmodulates the biological function of most of these chemokines and cytokines (Table 3). Consistent with this observation, it has been found that CD26 inhibitors can enhance some in vivo immune responses depending on the dose, application route, timing or predominant Teff subset [45, 169], which could be explained by the presence of off-target effects as well [25, 170]. However, animal models of rheumatoid arthritis [93], multiple sclerosis [171] and inflammatory bowel disease [172] in CD26 KO mice do support an immunosuppressive role of both CD26 and DPP4 activity. This immunosuppressive function has also been observed for sCD26 during strong in vitro proliferative responses of immune cells [143, 145]. Moreover, CD26 has been even regarded a tumour suppressor gene in melanomas or neuroblastomas [173, 174]. For this reason, we will focus this part of this review on the CD26 substrates with a direct connection with the immune system like chemokines.
CD26, TH2-Related Chemokines and TH2 high Asthma
Even though cytokines like IL-1β or IL-2 contain an appropriate N-terminal sequence, the molecular weight precludes their N-terminal clipping, with exceptions like IL-3, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF) or erythropoietin (Epo) [168]. In contrast, CD26-dependent processing of chemokines (8–10 kDa) like RANTES (regulated on activation, normal T cell expressed and secreted) is one of the most interesting aspects of CD26 biology [175]. Moreover, this function is mostly dependent on CD26 levels, which is influenced by the T cell subset or disease, as we have just seen.
Chemokines are proinflammatory cytokines with a role in leukocyte activation and migration. They are classified according to the position of two N-terminal cysteine residues (CC, CXC, C and CX3C) [176] but can also be divided as a function of the cells they attract or the receptor they recognize. As Table 3 shows, the major branches of effector (TH1, TH2 and TH17) and regulatory T cells express characteristic (but not totally selective) chemokine receptors [76, 103]. For example, TH2 lymphocytes and other leukocytes important in asthma (basophils, mast cells and eosinophils) express CCR3, CCR4, CCR8, CXCR4 and, in humans, CRTH2 [103]. CCR3, CCR4 and CXCR4 interact with CD26 substrates, but only CCR3 and CCR4 are actually specific for TH2 cells. Regarding CXCR4, TH2 cells seem to display a slight overexpression compared to TH1 lymphocytes [177], but other authors describe CXCR4 as a merely trafficking marker [178–180].
Ligation of CCR3 with eotaxin/CCL11; RANTES/CCL5; and MCP-1/CCL2, MCP-2/CCL8, MCP-3/CCL7 and MCP-4/CCL13 participates in the recruitment of basophils, eosinophils and mast cells [103, 181, 182] (Table 3). Eotaxin is an important chemokine in allergic asthma produced by endothelial cells and monocytes in response to IFNγ and TNFα, respectively. This chemokine is recognized by CCR3 (eosinophils, basophils, mast cells and TH2) and at lesser extent by CCR5, a putative TH1 marker (see later) [183]. Eotaxin is a chemoattractant for eosinophils that facilitates their mobilization from bone marrow [184]. Truncation of eotaxin by CD26 is characterized by an intermediate-low efficiency (kcat/Km: SDF1α > MDC > I-TAC > IP-10 > MIG > eotaxin > RANTES > LD78β) [185]. Despite this, N-terminal clipping by CD26 results in an isoform (3–74) with reduced chemotactic activity that causes CCR3 desensitization [183, 186, 187]. Indeed, administration of eotaxin to F344 rats leads to a mobilization of eosinophils, an effect enhanced in CD26-deficient animals or with CD26 inhibitors [187]. Additionally, CD26−/− mice challenged with ovalbumin (OVA) show higher levels of CCR3 and CCR3 ligands (eotaxin, RANTES) and stronger eosinophil infiltration in lungs [16]. Another ligand of CCR3 is CCL14 (Table 3), a chemokine processed by plasmin and urokinase plasminogen activator (UPA) to yield CCL14 (9–74). This isoform binds to CCR3 (and also CCR1 and CCR5) to efficiently attract eosinophils, monocytes and TH2 cells, a process also dampened by CD26 [188]. Therefore, a CD26−/low phenotype in both eosinophils and TH2 cells (CCR3+) seems to favour the inflammatory response in TH2high asthma.
CXCR4 is found in monocytes and B/T (preferentially TH2) cells [177, 189] and promotes the recruitment of T cells in lungs during allergic airway diseases [178, 190]. CXCR4 recognizes macrophage migration inhibitory factor (MIF) and stromal cell-derived factor 1 (SDF1/CXCL12) [189] (Table 3), the last one a small chemokine synthesized by endothelial cells and fibroblasts that induces T/B cell activation [142] and regulates the traffic of lymphocytes, monocytes and dendritic cells towards inflamed epithelia [179, 189, 191, 192]. Amongst the several SDF1 isoforms, at least two (SDF1α and SDF1β) are efficiently processed by CD26 in vitro [185, 193–195] and in vivo [93]. The SDF1α (3–67) isoform is unable to activate CXCR4 and displays antagonistic activity [185, 193–198]. Moreover, both CD26 and CXCR4 have similar expression kinetics upon activation [58, 199] and interact in lymphocytes [114]. Binding of SDF1α to CXCR4 initiates the CXCR4-CD26 complex endocytosis [114], a process disrupted by N-terminal clipping of SDF1α [195]. This finding could not explain however the preferential expression of CXCR4 in CD26low cells (e.g., TH2 or naïve TH) [76, 199] or why TGF-β (a cytokine that induces CD26 downmodulation) leads to augmented expression of CXCR4 and a potentiated SDF1α-CXCR4 axis [200–202]. This CD26−/lowCXCR4+ phenotype also facilitates a vigorous response to SDF1α in the recruitment of T cells and eosinophils in a mouse model of lung allergic inflammation [178]. On the other hand, CD26 has also been positively correlated with the in vitro invasive capacity of T cell lines in response to SDF1α, and this positive effect is mediated by CD45 [203], a tyrosine phosphatase that enhances cell migration in response to SDF1α [204].
CCR4 is present on monocytes, dendritic cells, NK cells and TH cells (TH2, TH17, Treg). CCR4 recognizes both CCL17/TARC and CCL22/MDC (Table 3), the last chemokine produced by macrophages, dendritic cells, NK cells and B/T lymphocytes [205]. TARC and MDC are expressed by epithelial cells in the airways, and their levels are upregulated after allergen challenge [103]. TH2 cytokines (e.g., IL-4 and IL-13), LPS, IL-1 and TNFα stimulate the secretion of MDC, whereas TH1 cytokines (e.g., IFNα, IL-12) inhibit MDC production [205, 206]. Moreover, MDC generates and amplifies TH2 responses and recruits TH2 lymphocytes [205–207], being important in diseases with a TH2-cytokine profile (e.g., asthma) or equivalent models in mice [207]. In these models, CCR4-MDC has a dominant role in later and chronic stages of the disease as compared with CCR3-eotaxin [181]. Furthermore, CD26 processes MDC (half-life 2–5 min) very efficiently to generate MDC(3–69) and, subsequently, MDC(5–69) [185]. This last isoform preserves the attractant power for monocytes, but displays reduced chemotactic activity for lymphocytes and dendritic cells [150], two subsets important in asthma. As Tregs, TH2 and CCR4+ TH17 cells are CD26−/low cells, while CCR4− TH17 cells are CD26high cells [72, 79], the CD26−/low phenotype of these CCR4+ subsets should apparently facilitate their recruitment to inflammatory sites.
CD26, TH1/TH17-Related Chemokines and TH2 low Asthma
Most of asthma patients present a TH2high disease, but there are other two types (obesity-associated and neutrophilic asthma) linked to a TH2low (i.e., TH1/TH17) profile [1]. The CD26high T cell subset displays a CD45RO+CCR7low effector-memory phenotype and produces TH1/TH17 cytokines [72, 77, 208]. TH1 cells are CCR5+CXCR3A+ lymphocytes [72, 208]; are essential for the production of IgM, IgG and IgA (but not IgE) by B cells; and orchestrate the response against intracellular microbes [103]. A few CCR5 ligands and all the CXCR3A-specific chemokines are CD26 substrates (Table 3). For example, CCL5/RANTES is produced by endothelial and epithelial cells, platelets, macrophages, eosinophils and T cells [209]. RANTES activates CCR1, CCR3 (TH2) and preferentially CCR5 (TH1), thereby attracting monocytes, eosinophils and T cells to inflammatory sites [209]. This chemokine is cleaved with a low efficiency (half-life of 400 min) by CD26 [100, 185], leading to RANTES(3–68), a chemokine that loses its capacity to bind CCR1 and CCR3 (monocytes, eosinophils and TH2 cells), but not CCR5 [197]. As CCR5 is expressed on TH1 and CD45RA−CD45R0+CCR7low effector-memory CD4+ T cells (both CD26+/high) [76, 103, 199], this means that RANTES(3–68) favours their attraction towards inflammatory sites [175, 197, 198, 210].
Another TH1 chemokine and CD26 substrate that binds CCR5 is LD78β/CCL3L1 [211]. Macrophage inflammatory protein-1α (MIP-1α) is encoded by two different loci: LD78α and LD78β [212]. Both chemokines are agonists of CCR1, CCR3 and CCR5, but LD78β binds with high affinity to CCR5 [213], regulating the traffic/activation of macrophages/monocytes, NK cells, eosinophils, basophils, immature dendritic cells and T lymphocytes. Strikingly, the inefficient (half-life ∼ 5 h) [185, 211] clipping of LD78β by CD26 generates LD78β(3–70) and enhances the chemotactic activity for TH1 cells (CCR5) and monocytes/neutrophils (CCR1) (Table 3). This fact, together with the CCR1low phenotype in eosinophils (<20 %) and the reduced affinity of LD78β (3–70) for CCR3 (TH2), proves once again that this posttranslational modification tends to favour TH1 responses [211, 214].
CXCR3A is overrepresented on TH1 cells [208] and is recognized by IFNγ-induced chemokines (CXCL9/Mig, CXCL10/IP-10, CXCL11/I-TAC) secreted by endothelial cells, hepatocytes, fibroblasts, keratinocytes and leucocytes in response to infections and several diseases including asthma. These chemokines are important to bring TH1 cells into epithelial barriers [103]. Moreover, after being N-terminally processed by CD26 with intermediate-high efficiency [185] (Table 3), they become less active TH1 chemoattractants. In addition, this modification also makes IP-10 a CXCR3 antagonist and induces receptor desensitization, being part of a negative feedback mechanism important to downmodulate inflammation [215–217].
TH17 lymphocytes play a major role in tissue inflammation, are essential to activate macrophages and recruit neutrophils and drive the defensive activities against extracellular bacteria and fungi. Human TH17 cells are associated with the expression of CCR4, CCR6 and CXCR6 [72, 103, 218]. Skin-homing addressins (e.g., CCR4, CCR6) are also shared by CD26−/low Treg cells and TH2 cells [73]. However, TH17 cells are rare at sites of inflammation, maybe due to homeostatic mechanisms that avoid their expansion, a high plasticity to shift to TH1 cells [103] or the heterogeneous expression of CCR4, with a proinflammatory CCR4−CCR6+CD26high phenotype in most of TH17 cells and a small subset of immunosuppressive CCR4+CCR6+CD26low [79, 81]. This heterogeneity for CCR4 is also detected in TH22 cells, a lineage that produces IL-22; expresses CLA, CCR6 and CCR10; and plays an anti-inflammatory and tissue-protective role in asthma [219]. Curiously, CCR4− TH22 cells are CD26−, whereas CCR4+ TH22 cells are constituted by either CD26− or CD26+ lymphocytes [79].
In summary, taking into consideration the CD26 expression gradient in the major TH subsets (TH17 >> TH1 > TH2>> Treg), the small proportion of chemokines affecting Treg/TH2/TH17 cells that are processed by CD26 as compared with those recruiting TH1 cells and that CD26-dependent clipping produces a drop in the biological functions of most of them, it is evident that CD26 forms part of a homeostatic mechanism to downmodulate airway inflammation mediated by TH2 and especially TH17/TH1 cells (e.g., obesity-linked asthma).
CD26 and Asthma: Animal Models, Studies in Human System and Clinical Implications
Despite the reported evidences on a role of CD26 in TH1-related diseases, the involvement of both the peptidase-dependent and peptidase-independent functions of CD26 in the pathophysiology of asthma is not yet fully understood. In this section, we summarize some results obtained in animal models and the human system.
Rat Models
Rat models using inhaled allergens such as OVA have been used to evaluate the role of CD26 in bronchial asthma. Thus, the severity of the airway inflammation decreases as the endogenous CD26 level of the strain becomes lower [220], likely due to the costimulatory role of CD26. One of the models more extensively used is the F344 strain. Challenge of F344 rats with OVA generates bronchoconstriction and higher CD26 expression on lymphocytes and epithelial cells of lung parenchyma [14, 17]; enhanced levels of DPP8/9 and DPP10 in bronchi [17]; and a dose-dependent recruitment to the lungs of dendritic cells, eosinophils and CD4+CD25+CD26+ T cells [14]. More specifically, OVA causes increased recruitment of T cells to bronchi (but not lung parenchyma) [22] and enhanced sDPP4 activity in bronchoalveolar lavage fluid (BALF) [17], the last finding likely related with the augmented presence of CD26− T cells in bronchi. Interestingly, there is a F344 substrain with an active-site mutation in the Dpp4 gene that causes protein retention/degradation in the endoplasmic reticulum and lower CD26 levels [221]. This F344 substrain also displays the early bronchoconstriction in response to OVA challenge [222], but the late inflammatory response is milder regarding OVA-specific serum IgE, eosinophilia and recruitment of T cells in both bronchi and BALF [14, 22, 220]. In contrast, entrance of T cells in bronchoalveolar lymphoid tissue (BALT) is enhanced, no matter the CD26+ or CD26− phenotype of transferred T cells [223].
Because bronchi are DPP4− (but DPP8/9/10+) compartments [17], CD4+CD25+ T cells recruited in the airways of OVA-challenged F344 rats are either TH cells with a CD26low phenotype or they downmodulate CD26 during peribronchial infiltration. Thus, a potential higher response of CD4+CD25+CD26+ T cells to SDF1α [203] could allow their recruitment to bronchi (CD26− SDF-1high environment) of wild-type rats [22] with a concomitant SDF1-driven downmodulation of CD26, as it happens during skin homing of Sézari cells [224]. In contrast, CD4+CD25+CD26− Teff cells in CD26-deficient rats would not be attracted to the bronchi as a result from either an anomalous/aberrant response to SDF1α [203] or a reduced chemokine gradient [22], resulting in a milder asthma-like disease. However, despite that SDF1 is augmented in the bronchi of asthmatic patients [225], OVA challenge does not increase the number of SDF1 transcripts in the large airways of F344 rats [22]. Curiously, the amount of SDF1 is increased in the BALT of CD26-deficient rats, likely supporting the above commented higher recruitment of T cells to this lymphoid compartment after OVA challenge [223].
CD26-deficient F344 rats display a parallel increased influx of Tregs into the lungs, with a concomitant raise in the IL-10 production that explains the diminished inflammation [222]. However, this may not happen in the same way in humans, where Treg cells display a CD26−/low phenotype [69–72] in contrast to rats [14, 68]. Moreover, it is difficult to conciliate a milder asthmatic-like inflammation in OVA-challenged CD26-deficient F344 rats with either a lower truncation rate of TH2 chemokines (e.g., eotaxin) or the in vivo enhanced eotaxin-mediated recruitment of eosinophils caused by a CD26 inhibitor [187]. Perhaps the different genetic background [226] or the administration route might play a significant role [45]. For example, oral administrated inhibitors seem to enhance allergic inflammation of the airways, whereas topic inhibition (aerosolization) has a rather protective effect in line with that observed in CD26-deficient F344 rats [45]. Moreover, dual deficiency of enzymatic and extraenzymatic activities of CD26 in murine asthma models may generate results different from the observed effects of CD26 inhibitors, which only inhibit enzymatic functions [166].
Mouse Models
CD26 KO mice display a healthy phenotype, with increased glucose clearance, resistance to obesity and small changes in the percentages of NK, NKT and CD4+ T cells [227, 228]. In addition, in vitro pokeweed mitogen (PWM)-activated splenocytes from CD26−/− C56BL/6 mice show a decreased IL-4 production, while IgE and IL-4 are significantly reduced in sera from CD26−/− animals upon PWM treatment [228]. In clear contrast, Yan et al. reported in 2012 unchanged IgE concentration but enhanced eosinophilia and TH2 cytokines (IL-4, IL-5, IL-13) in BALF from OVA-induced CD26−/− C56BL/6 animals, as well as higher mRNA and protein levels of CD26 substrates (eotaxin and RANTES) and chemokine receptors (CCR3 and CCR5) [16]. These partially contradictory results probably depend on the type, administration route and strength of the polyclonal stimulus (PWM vs. OVA) [16]. They also point out that induced and targeted (cell type-specific) KO models could be necessary to truly dissect the role of CD26 in asthma [16].
Human System
In line with the small differences in CD26 levels observed between TH1 and TH2 cells [72], this marker is not helpful to differentiate TH1- from TH2-driven responses in patients with atopic asthma [229]. However, augmented CD26 levels are actually detected on total lymphocytes, CD4+ T cells and iNKT (but not CD8+ T lymphocytes, monocytes or B cells) in adult allergic asthma, reflecting the existence of an activated status [20]. This last work also published an elevation of plasma sCD26 in patients, which was associated with eosinophil counts and IgE concentration. Additionally, this group reported a lower production of TH1 chemokines (IP-10, MIG) and higher presence of other chemokines (RANTES, MDC) and their receptors (CCR3, CCR4), in both cases attributable to a TH2-response predominance [20]. In clear contrast, Matsuno et al. found that sCD26 is inversely correlated with the inflammation level in chronic eosinophilic pneumonia, a disease frequently preceded or accompanied by asthma [230]. More recently, no differences were found in serum sCD26 levels in children with asthma or association of this variable with either asthma or atopy (e.g., skin prick test, IgE, eosinophil cationic protein) [86].
From the analysis of all these studies, the need to expand them to different asthmatic phenotypes (or endotypes) and levels of severity/chronicity is obvious. In addition, it is also clear that other variables (sample size, lymphocytes counts, age, sex) should also be taken into account, especially knowing (a) the influence of sex on membrane-bound CD26 (our unpublished results) and sCD26 levels [86–89, 91] and (b) the augmented prevalence of asthma in boys at early ages and the higher prevalence of asthma in women after puberty [5, 6], especially in the case of certain TH2low phenotypes [1]. For example, sCD26 is apparently increased in the blood of both atopic dermatitis (a TH2-like disease) [231] and adult asthma patients [20], but in the last study these results might be partially explained by a higher proportion of males in the group of patients. In contrast, Remes did not show altered sCD26 levels in asthmatic children and took into consideration certain pre-analytical variables (gender, age) [86]. Therefore, more studies are needed to reveal the potential correlation between CD26/sCD26 and the severity and phenotype of the asthmatic pathology.
Clinical Implications
High body mass index is a factor with a positive association with asthma development and severity [3, 232, 233]. This association of asthma and obesity appears to be stronger in women than in men, with a role for both non-inflammatory and subclinical/systemic chronic inflammatory pathways, including the release of proinflammatory (e.g., leptin) or anti-inflammatory (e.g., adiponectin) adipokines that regulate the survival of eosinophils and their recruitment to the lungs [232, 233]. As mentioned above, visceral fat in obese patients releases the “adipokine” sCD26 into the circulation [88], but, as we have already been highlighting throughout this review, with proinflammatory or anti-inflammatory effects depending on the processed substrate: incretins or chemokines/substance P, respectively.
Obese persons are at risk of a number of comorbidities like type 2 diabetes mellitus (T2DM), a disease characterized by impaired production of incretins, with subsequently hyperglycaemia. Incretins (GIP, GLP-1, GLP-2) are well-known CD26 substrates that lose their function when they are processed. Inhibition of CD26 enzymatic activity avoids the degradation of these hormones, enhances the incretin effect, improves both the insulin secretion and the glucose uptake and lowers the glycosylated haemoglobin A1c levels. To achieve these goals, a new group of anti-hyperglycaemic drugs (CD26 inhibitors or gliptins) have come on the market, with sitagliptin, (Januvia®; Merck Sharp & Dohme Ltd) and vildagliptin (Galvus®; Novartis Europharm Ltd) (both approved by European Medicines Agency/EMA in 2007) as the first outpost (Table 4). Currently, there are several orally administered CD26 inhibitors that have been approved by agencies like the Food and Drug Administration (FDA) or EMA and are being used with satisfactory results as a second- or third-line medication in combination with other oral anti-diabetic drugs. Commercially available gliptins include the above-mentioned sitagliptin (Januvia®, Ristaben®) but also saxagliptin (Onglyza®), linagliptin (Trajenta®), alogliptin (Vipidia®) and vildagliptin (Galvus®, Jalra®, Xiliarx®) (Table 4). However, there is a controversial debate about the secondary effects on comorbidities (e.g., cardiovascular outcome, renal impairment, acute pancreatitis) that the long-term treatment with these long half-life inhibitors may have. These concerns are a result of several issues related to the great number of CD26 substrates linked or not to the immune system. Thus, the SAVOR TIMI-53 (saxagliptin) [234] trial detected a significantly increased rate for hospitalization due to heart failure, and a more recent study based on FAERS (US-FDA Adverse Event Reporting System) [235] is also consistent with this finding. In clear contrast, the TECOS trial (sitagliptin) [236] did not find an increased risk of heart failure.
More in accordance with the scope of this review, CD26 reversible inhibitors seem to increase the frequency of risk factors for asthma development/exacerbation like atopic sensitization, rhinitis or rhinovirus infection in T2DM patients [3]. Thus, a rapid review of clinical data published on the EMA web page (http://www.ema.europa.eu) supports increased risk of non-serious upper respiratory tract infections (e.g., viral nasopharyngitis, rhinitis, sinusitis) (Table 4), which would be in line with the costimulatory role of CD26. As Willemen and coauthors sustain, this increased risk of infections with CD26 inhibitors (∼3 % cases) cannot be compared with the magnitude of the effects seen with biological agents like tumour necrosis factor inhibitors [237]. However, this effect should not be underestimated and must be added to the immune impairment caused by T2DM itself to increase the overall asthma development/exacerbation risk in these patients.
Other adverse drug reactions (ADRs) reported in association with gliptins and more associated with an inhibitory role of CD26 would be interstitial lung disease, hypersensitivity reactions (including anaphylactic responses or bronchial hyperreactivity) and skin and subcutaneous tissue disorders such as pruritus, angioedema, rash, urticaria, cutaneous vasculitis or more severe and rare skin conditions (Table 4). For example, Bullous pemphigoid [187, 238–240] is an autoimmune subepidermal disease with overproduction of eotaxin and eosinophilia that affects skin and mucosae. Therefore, the association between gliptins and bullous pemphigoid is in tune with in vivo studies in F344 rats showing that CD26 limits the eotaxin-mediated recruitment of eosinophils [187] or with the finding that oral administration of CD26 inhibitors promotes the allergic inflammation of the airways [45]. This means that CD26 could be part of a homeostatic mechanism to downmodulate airway inflammation mediated by TH2 and especially TH17/TH1 cells important in some TH2low asthma phenotypes such as obesity-related asthma. Nevertheless, it should be noted that besides chemokines there are other CD26 substrates with implications on obesity and asthma like substance P. Inhaled substance P, but not bradykinin, enhances the airway response to bronchoconstricting agents in guinea pigs [241]. Substance P also favours allergen sensitization and bronchial inflammation, as observed in a mouse model of diet-induced obesity sensitized and challenged with OVA and treated with an antagonist of the substance P receptor (NK1-R) [242]. Therefore, expanded half-life in substance P caused by treatment with CD26 inhibitors in T2DM patients could increase certain obesity-related comorbidities such as asthma. In the same way, administration of sitagliptin in T2DM generates a temporary decrease in the percentage of peripheral blood Tregs that also could favour asthma prevalence in these patients [243]. In clear contrast, GLP-1-based therapies have anti-inflammatory effects in chronic inflammatory diseases including T2DM and asthma [244]. For example, in vitro studies have described that CD26 inhibitors induce a decrease of NLRP3 inflammasome, toll-like receptor 4 (TLR4) signalling and IL-1β in macrophages via GLP-1 receptor [160]. Therefore, more studies are needed to clarify whether gliptins have an overall positive or negative role in T2DM patients with obesity-related asthma.
Concluding Remarks
CD26 is a relevant molecule in asthma for several reasons. The first one is because of its potential utility together with periostin to test the efficacy of the treatment with interleukin-13-neutralising monoclonal antibodies (e.g., Tralokinumab) in patients with severe uncontrolled asthma [245, 246]. Secondly, it is because of its multifunctionality nature, with both costimulatory and inhibitory roles in the immune system, apart from the high (but variable) CD26 levels in a subset of lymphocytes central to asthma: the CD4+ T cells. Different authors have highlighted the inhibitory functions of this molecule relative to signalling mediated by cytokines and chemokines. A handful of chemokines important for CD26+ TH2 cells during early-onset allergic or late-onset eosinophilic asthma includes chemotactic factors whose biological activity is reduced with low-intermediate (eotaxin) or high (MDC, SDF1α) efficiency, whereas important chemokines for CD26+ TH1 (I-TAC, IP-10, Mig) and CD26high TH17 cells (MDC), two Teff subsets relevant in obesity-related asthma, late-onset neutrophilic asthma or severe asthma, are inactivated with intermediate-high effectiveness. Therefore, CD26 could act as a “biological brake” by reducing the attraction of Teff cells at sites of inflammation (e.g., the bronchi). This means that CD26 inhibitors used in T2DM patients could lead to a higher persistence of unprocessed substance P or chemokines and exacerbation of TH2high and especially TH2low asthma. In conclusion, it is necessary to previously take into consideration the many existing questions regarding the biological functions of this enzyme before the therapeutic targeting of this molecule [247], as CD26 inhibitors could enhance the frequency of hospital admission amongst people with certain asthma phenotypes.
References
Wenzel SE (2012) Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 18:716–725
Holgate ST (2012) Innate and adaptive immune responses in asthma. Nat Med 18:673–683
Beasley R, Semprini A, Mitchell EA (2015) Risk factors for asthma: is prevention possible? Lancet 386:1075–1085
Moorman JE, Akinbami LJ, Bailey CM, Zahran HS, King ME, Johnson CA, Liu X (2012) National surveillance of asthma: United States, 2001–2010. National Center for Health Statistics. Vital Health Stat 3(35):1–58
de Marco R, Locatelli F, Sunyer J, Burney P (2000) Differences in incidence of reported asthma related to age in men and women. A retrospective analysis of the data of the European Respiratory Health Survey. Am J Respir Crit Care Med 162:68–74
Tan DJ, Walters EH, Perret JL, Lodge CJ, Lowe AJ, Matheson MC, Dharmage SC (2015) Age-of-asthma onset as a determinant of different asthma phenotypes in adults: a systematic review and meta-analysis of the literature. Expert Rev Respir Med 9:109–123
Mathias RA, Grant AV, Rafaels N, Hand T, Gao L, Vergara C, Tsai YJ, Yang M, Campbell M, Foster C, Gao P, Togias A, Hansel NN, Diette G, Adkinson NF, Liu MC, Faruque M, Dunston GM, Watson HR, Bracken MB, Hoh J, Maul P, Maul T, Jedlicka AE, Murray T, Hetmanski JB, Ashworth R, Ongaco CM, Hetrick KN, Doheny KF, Pugh EW, Rotimi CN, Ford J, Eng C, Burchard EG, Sleiman PM, Hakonarson H, Forno E, Raby BA, Weiss ST, Scott AF, Kabesch M, Liang L, Abecasis G, Moffatt MF, Cookson WO, Ruczinski I, Beaty TH, Barnes KC (2010) A genome-wide association study on African-ancestry populations for asthma. J Allergy Clin Immunol 125:336–346
Portelli M, Sayers I (2012) Genetic basis for personalized medicine in asthma. Expert Rev Respir Med 6:223–236
Martinez FD, Vercelli D (2013) Asthma. Lancet 382:1360–1372
Mentlein R (1999) Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul Pept 85:9–24
Heutinck KM, ten Berge IJ, Hack CE, Hamann J, Rowshani AT (2010) Serine proteases of the human immune system in health and disease. Mol Immunol 47:1943–1955
Qi SY, Riviere PJ, Trojnar J, Junien JL, Akinsanya KO (2003) Cloning and characterization of dipeptidyl peptidase 10, a new member of an emerging subgroup of serine proteases. Biochem J 373:179–189
Allen M, Heinzmann A, Noguchi E, Abecasis G, Broxholme J, Ponting CP, Bhattacharyya S, Tinsley J, Zhang Y, Holt R, Jones EY, Lench N, Carey A, Jones H, Dickens NJ, Dimon C, Nicholls R, Baker C, Xue L, Townsend E, Kabesch M, Weiland SK, Carr D, von Mutius E, Adcock IM, Barnes PJ, Lathrop GM, Edwards M, Moffatt MF, Cookson WO (2003) Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet 35:258–263
Skripuletz T, Schmiedl A, Schade J, Bedoui S, Glaab T, Pabst R, von Hörsten S, Stephan M (2007) Dose-dependent recruitment of CD25+ and CD26+ T cells in a novel F344 rat model of asthma. Am J Physiol Lung Cell Mol Physiol 292:L1564–L1571
Zhou H, Hong X, Jiang S, Dong H, Xu X, Xu X (2009) Analyses of associations between three positionally cloned asthma candidate genes and asthma or asthma-related phenotypes in a Chinese population. BMC Med Genet 10:123–131
Yan S, Gessner R, Dietel C, Schmiedek U, Fan H (2012) Enhanced ovalbumin-induced airway inflammation in CD26−/− mice. Eur J Immunol 42:533–540
Schade J, Stephan M, Schmiedl A, Wagner L, Niestroj AJ, Demuth HU, Frerker N, Klemann C, Raber KA, Pabst R, von Hörsten S (2008) Regulation of expression and function of dipeptidyl peptidase 4 (DP4), DP8/9, and DP10 in allergic responses of the lung in rats. J Histochem Cytochem 56:147–155
Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF (2013) Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500:227–231
Bønnelykke K, Vissing NH, Sevelsted A, Johnston SL, Bisgaard H (2015) Association between respiratory infections in early life and later asthma is independent of virus type. J Allergy Clin Immunol 136:81–86
Lun SW, Wong CK, Ko FW, Hui DS, Lam CW (2007) Increased expression of plasma and CD4+ T lymphocyte costimulatory molecule CD26 in adult patients with allergic asthma. J Clin Immunol 27:430–437
Rojas-Ramos E, Garfias Y, Jiménez-Martínez Mdel C, Martínez-Jiménez N, Zenteno E, Gorocica P, Lascurain R (2007) Increased expression of CD30 and CD57 molecules on CD4(+) T cells from children with atopic asthma: a preliminary report. Allergy Asthma Proc 28:659–666
Schade J, Schmiedl A, Kehlen A, Veres TZ, Stephan M, Pabst R, von Hörsten S (2009) Airway-specific recruitment of T cells is reduced in a CD26-deficient F344 rat substrain. Clin Exp Immunol 158:133–142
Torimoto Y, Dang NH, Tanaka T, Prado C, Schlossman SF, Morimoto C (1992) Biochemical characterization of CD26 (dipeptidyl peptidase IV): functional comparison of distinct epitopes recognized by various anti-CD26 monoclonal antibodies. Mol Immunol 29:183–192
De Meester I, Vanhoof G, Hendriks D, Demuth HU, Yaron A, Scharpé S (1992) Characterization of dipeptidyl peptidase IV (CD26) from human lymphocytes. Clin Chim Acta 210:23–34
Gorrell MD, Gysbers V, McCaughan GW (2001) CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol 54:249–264
Ajami K, Abbott CA, Obradovic M, Gysbers V, Kähne T, McCaughan GW, Gorrell MD (2003) Structural requirements for catalysis, expression, and dimerization in the CD26/DPIV gene family. Biochemistry 42:694–701
Chien CH, Tsai CH, Lin CH, Chou CY, Chen X (2006) Identification of hydrophobic residues critical for DPP-IV dimerization. Biochemistry 45:7006–7012
Jascur T, Matter K, Hauri HP (1991) Oligomerization and intracellular protein transport: dimerization of intestinal dipeptidylpeptidase IV occurs in the Golgi apparatus. Biochemistry 30:1908–1915
Danielsen EM (1994) Dimeric assembly of enterocyte brush border enzymes. Biochemistry 33:1599–1605
Fleischer B (1994) CD26: a surface protease involved in T-cell activation. Immunol Today 15:180–184
Morimoto C, Schlossman SF (1998) The structure and function of CD26 in the T-cell immune response. Immunol Rev 161:55–70
Dobers J, Grams S, Reutter W, Fan H (2000) Roles of cysteines in rat dipeptidyl peptidase IV/CD26 in processing and proteolytic activity. Eur J Biochem 267:5093–5100
Ohnuma K, Dang NH, Morimoto C (2008) Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol 29:295–301
Chung KM, Huang CH, Cheng JH, Tsai CH, Suen CS, Hwang MJ, Chen X (2011) Proline in transmembrane domain of type II protein DPP-IV governs its translocation behavior through endoplasmic reticulum. Biochemistry 50:7909–7918
Dinjens WN, Ten Kate J, Kirch JA, Tanke HJ, Van der Linden EP, Van den Ingh HF, Van Steenbrugge GJ, Meera Khan P, Bosman FT (1990) Adenosine deaminase complexing protein (ADCP) expression and metastatic potential in prostatic adenocarcinomas. J Pathol 160:195–201
Abbott CA, Baker E, Sutherland GR, McCaughan GW (1994) Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 40:331–338
Wesley UV, Albino AP, Tiwari S, Houghton AN (1999) A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J Exp Med 190:311–322
Wesley UV, Tiwari S, Houghton AN (2004) Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int J Cancer 109:855–866
Wesley UV, McGroarty M, Homoyouni A (2005) Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res 65:1325–1334
van der Velden VH, Wierenga-Wolf AF, Adriaansen-Soeting PW, Overbeek SE, Möller GM, Hoogsteden HC, Versnel MA (1998) Expression of aminopeptidase N and dipeptidyl peptidase IV in the healthy and asthmatic bronchus. Clin Exp Allergy 28:110–120
De Meester I, Korom S, Van Damme J, Scharpé S (1999) CD26, let it cut or cut it down. Immunol Today 20:367–375
van der Velden VH, Hulsmann AR (1999) Peptidases: structure, function and modulation of peptide-mediated effects in the human lung. Clin Exp Allergy 29:445–456
Hong WJ, Petell JK, Swank D, Sanford J, Hixson DC, Doyle D (1989) Expression of dipeptidyl peptidase IV in rat tissues is mainly regulated at the mRNA levels. Exp Cell Res 182:256–266
Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC, Erle DJ (2007) IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 36:244–253
Stephan M, Suhling H, Schade J, Wittlake M, Tasic T, Klemann C, Pabst R, Jurawitz MC, Raber KA, Hoymann HG, Braun A, Glaab T, Hoffmann T, Schmiedl A, von Hörsten S (2013) Effects of dipeptidyl peptidase-4 inhibition in an animal model of experimental asthma: a matter of dose, route, and time. Physiol Rep 1:e00095
Micouin A, Bauvois B (1997) Expression of dipeptidylpeptidase IV (DPP IV/CD26) activity on human myeloid and B lineage cells, and cell growth suppression by the inhibition of DPP IV activity. Adv Exp Med Biol 421:201–205
Yamabe T, Takakura K, Sugie K, Kitaoka Y, Takeda S, Okubo Y, Teshigawara K, Yodoi J, Hori T (1997) Induction of the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural killer cells by IL-2, IL-12 or IL-15. Immunology 91:151–158
von Bonin A, Hühn J, Fleischer B (1998) Dipeptidyl-peptidase IV/CD26 on T cells: analysis of an alternative T-cell activation pathway. Immunol Rev 161:43–53
Dang NH, Torimoto Y, Sugita K, Daley JF, Schow P, Prado C, Schlossman SF, Morimoto C (1990) Cell surface modulation of CD26 by anti-1F7 monoclonal antibody. Analysis of surface expression and human T cell activation. J Immunol 145:3963–3971
Mattern T, Reich C, Duchrow M, Ansorge S, Ulmer AJ, Flad HD (1995) Antibody-induced modulation of CD26 surface expression. Immunology 84:595–600
Kähne T, Kröning H, Thiel U, Ulmer AJ, Flad HD, Ansorge S (1996) Alterations in structure and cellular localization of molecular forms of DP IV/CD26 during T cell activation. Cell Immunol 170:63–70
Darmoul D, Baricault L, Sapin C, Chantret I, Trugnan G, Rousset M (1991) Decrease of mRNA levels and biosynthesis of sucrase-isomaltase but not dipeptidylpeptidase IV in forskolin or monensin-treated Caco-2 cells. Experientia 47:1211–1215
Kahne T, Lendeckel U, Wrenger S, Neubert K, Ansorge S, Reinhold D (1999) Dipeptidyl peptidase IV: a cell surface peptidase involved in regulating T cell growth. Int J Mol Med 4:3–15
Salgado FJ, Vela E, Martín M, Franco R, Nogueira M, Cordero OJ (2000) Mechanisms of CD26/dipeptidyl peptidase IV cytokine-dependent regulation on human activated lymphocytes. Cytokine 12:1136–1141
Rogge L, Bianchi E, Biffi M, Bono E, Chang SY, Alexander H, Santini C, Ferrari G, Sinigaglia L, Seiler M, Neeb M, Mous J, Sinigaglia F, Certa U (2000) Transcript imaging of the development of human T helper cells using oligonucleotide arrays. Nat Genet 25:96–101
Stefanovic V, Ardaillou N, Vlahovic P, Placier S, Ronco P, Ardaillou R (1993) Interferon-gamma induces dipeptidylpeptidase IV expression in human glomerular epithelial cells. Immunology 80:465–470
Bauvois B, Djavaheri-Mergny M, Rouillard D, Dumont J, Wietzerbin J (2000) Regulation of CD26/DPPIV gene expression by interferons and retinoic acid in tumor B cells. Oncogene 19:265–272
Cordero OJ, Salgado FJ, Viñuela JE, Nogueira M (1997) Interleukin-12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology 197:522–533
Cordero OJ, Salgado FJ, Viñuela JE, Nogueira M (1998) Interleukin-12-dependent activation of human lymphocyte subsets. Immunol Lett 61:7–13
Bühling F, Kunz D, Reinhold D, Ulmer AJ, Ernst M, Flad HD, Ansorge S (1994) Expression and functional role of dipeptidyl peptidase IV (CD26) on human natural killer cells. Nat Immun 13:270–279
Bühling F, Junker U, Reinhold D, Neubert K, Jäger L, Ansörge S (1995) Functional role of CD26 on human B lymphocytes. Immunol Lett 45:47–51
Bauvois B, De Meester I, Dumont J, Rouillard D, Zhao HX, Bosmans E (1999) Constitutive expression of CD26/dipeptidylpeptidase IV on peripheral blood B lymphocytes of patients with B chronic lymphocytic leukaemia. Br J Cancer 79:1042–1048
Cordero OJ, Salgado FJ, Fernández-Alonso CM, Herrera C, Lluis C, Franco R, Nogueira M (2001) Cytokines regulate membrane adenosine deaminase on human activated lymphocytes. J Leukoc Biol 70:920–930
Arndt M, Lendeckel U, Spiess A, Faust J, Neubert K, Reinhold D, Ansorge S (2000) Dipeptidyl peptidase IV (DP IV/CD26) mRNA expression in PWM-stimulated T-cells is suppressed by specific DP IV inhibition, an effect mediated by TGF-beta(1). Biochem Biophys Res Commun 274:410–414
Uematsu T, Tanaka H, Yamaoka M, Furusawa K (2004) Effects of oral squamous cell carcinoma-derived TGF-beta1 on CD26/DPPIV expression in T cells. Anticancer Res 24:619–624
Tan EY, Richard CL, Zhang H, Hoskin DW, Blay J (2006) Adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing ERK1/2 activity via a novel pathway. Am J Physiol Cell Physiol 29:C433–C444
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500
Stephens LA, Barclay AN, Mason D (2004) Phenotypic characterization of regulatory CD4 + CD25+ T cells in rats. Int Immunol 16:365–375
Kattah MG, Coller J, Cheung RK, Oshidary N, Utz PJ (2008) HIT: a versatile proteomics platform for multianalyte phenotyping of cytokines, intracellular proteins and surface molecules. Nat Med 14:1284–1289
Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, Lang S, Jackson EK, Gorelik E, Whiteside TL (2010) Generation and accumulation of immunosuppressive adenosine by human CD4 + CD25highFOXP3+ regulatory T cells. J Biol Chem 285:7176–7186
Salgado FJ, Pérez-Díaz A, Villanueva NM, Lamas O, Arias P, Nogueira M (2012) CD26: a negative selection marker for human Treg cells. Cytometry A 81:843–855
Bengsch B, Seigel B, Flecken T, Wolanski J, Blum HE, Thimme R (2012) Human Th17 cells express high levels of enzymatically active dipeptidylpeptidase IV (CD26). J Immunol 188:5438–5447
Clark RA, Kupper TS (2007) IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood 109:194–202
Romagnani S, Manetti R, Annunziato F, Maggi E (1995) Role of IL12 in the development of human Th1 type cells. Res Immunol 146:452–460
Willheim M, Ebner C, Baier K, Kern W, Schrattbauer K, Thien R, Kraft D, Breiteneder H, Reinisch W, Scheiner O (1997) Cell surface characterization of T lymphocytes and allergen-specific T cell clones: correlation of CD26 expression with T(H1) subsets. J Allergy Clin Immunol 100:348–355
Annunziato F, Galli G, Cosmi L, Romagnani P, Manetti R, Maggi E, Romagnani S (1998) Molecules associated with human Th1 or Th2 cells. Eur Cytokine Netw 9:12–16
Krakauer M, Sorensen PS, Sellebjerg F (2006) CD4(+) memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis. J Neuroimmunol 181:157–164
Zhao F, Hoechst B, Gamrekelashvili J, Ormandy LA, Voigtländer T, Wedemeyer H, Ylaya K, Wang XW, Hewitt SM, Manns MP, Korangy F, Greten TF (2012) Human CCR4+ CCR6+ Th17 cells suppress autologous CD8+ T cell responses. J Immunol 188:6055–6062
Cordero OJ, Varela-Calviño R, López-González T, Calviño-Sampedro C, Viñuela JE, Mouriño C, Hernández-Rodríguez Í, Rodríguez-López M, Aspe de la Iglesia B, Pego JM (2015) CD26 expression on T helper populations and sCD26 serum levels in patients with rheumatoid arthritis. PLoS One 10:e0131992
Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ (2012) Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 119:4430–4440
Ma Y, Visser L, Blokzijl T, Harms G, Atayar C, Poppema S, van den Berg A (2008) The CD4 + CD26- T-cell population in classical Hodgkin’s lymphoma displays a distinctive regulatory T-cell profile. Lab Invest 88:482–490
Cordero OJ, Salgado FJ, Nogueira M (2009) On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol Immunother 58:1723–1747
Van Der Velden VH, Naber BA, Van Hal PT, Overbeek SE, Hoogsteden HC, Versnel MA (1999) Peptidase activities in serum and bronchoalveolar lavage fluid from allergic asthmatics—comparison with healthy non-smokers and smokers and effects of inhaled glucocorticoids. Clin Exp Allergy 29:813–823
Durinx C, Lambeir AM, Bosmans E, Falmagne JB, Berghmans R, Haemers A, Scharpé S, De Meester I (2000) Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur J Biochem 267:5608–5613
Iwaki-Egawa S, Watanabe Y, Kikuya Y, Fujimoto Y (1998) Dipeptidyl peptidase IV from human serum: purification, characterization, and N-terminal amino acid sequence. J Biochem 124:428–433
Remes ST, Delezuch W, Pulkki K, Pekkanen J, Korppi M, Matinlauri IH (2011) Association of serum-soluble CD26 and CD30 levels with asthma, lung function and bronchial hyper-responsiveness at school age. Acta Paediatr 100:e106–e111
Durinx C, Neels H, Van der Auwera JC, Naelaerts K, Scharpe S, De Meester I (2001) Reference values for plasma dipeptidyl-peptidase IV activity and their association with other laboratory parameters. Clin Chem Lab Med 39:155–159
Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, Eckardt K, Kaufman JM, Ryden M, Müller S, Hanisch FG, Ruige J, Arner P, Sell H, Eckel J (2011) Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes 60:1917–1925
Delezuch W, Marttinen P, Kokki H, Heikkinen M, Vanamo K, Pulkki K, Matinlauri I (2012) Serum and CSF soluble CD26 and CD30 concentrations in healthy pediatric surgical outpatients. Tissue Antigens 80:368–375
De Chiara L, Rodríguez-Piñeiro AM, Cordero OJ, Rodríguez-Berrocal FJ, Ayude D, Rivas-Hervada And FJ, de la Cadena MP (2009) Soluble CD26 levels and its association to epidemiologic parameters in a sample population. Dis Markers 27:311–316
Tejera-Alhambra M, Casrouge A, de Andrés C, Ramos-Medina R, Alonso B, Vega J, Albert ML, Sánchez-Ramón S (2014) Low DPP4 expression and activity in multiple sclerosis. Clin Immunol 150:170–183
Kobayashi H, Hosono O, Mimori T, Kawasaki H, Dang NH, Tanaka H, Morimoto C (2002) Reduction of serum soluble CD26/dipeptidyl peptidase IV enzyme activity and its correlation with disease activity in systemic lupus erythematosus. J Rheumatol 29:1858–1866
Busso N, Wagtmann N, Herling C, Chobaz-Péclat V, Bischof-Delaloye A, So A, Grouzmann E (2005) Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. Am J Pathol 166:433–442
Duke-Cohan JS, Morimoto C, Rocker JA, Schlossman SF (1995) A novel form of dipeptidylpeptidase IV found in human serum. Isolation, characterization, and comparison with T lymphocyte membrane dipeptidylpeptidase IV (CD26). J Biol Chem 270:14107–14114
Duke-Cohan JS, Morimoto C, Rocker JA, Schlossman SF (1996) Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J Immunol 156:1714–1721
Duke-Cohan JS, Gu J, McLaughlin DF, Xu Y, Freeman GJ, Schlossman SF (1998) Attractin (DPPT-L), a member of the CUB family of cell adhesion and guidance proteins, is secreted by activated human T lymphocytes and modulates immune cell interactions. Proc Natl Acad Sci U S A 95:11336–11341
Wjst M (2007) Public data mining shows extended linkage disequilibrium around ADAM33. Allergy 62:444–446
Röhrborn D, Eckel J, Sell H (2014) Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett 588:3870–3877
Aso Y, Terasawa T, Kato K, Jojima T, Suzuki K, Iijima T, Kawagoe Y, Mikami S, Kubota Y, Inukai T, Kasai K (2013) The serum level of soluble CD26/dipeptidyl peptidase 4 increases in response to acute hyperglycemia after an oral glucose load in healthy subjects: association with high-molecular weight adiponectin and hepatic enzymes. Transl Res 162:309–316
Lambeir AM, Durinx C, Scharpé S, De Meester I (2003) Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci 40:209–294
Wang Z, Grigo C, Steinbeck J, von Hörsten S, Amann K, Daniel C (2014) Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides 57:109–117
Uematsu T, Urade M, Yamaoka M (1998) Decreased expression and release of dipeptidyl peptidase IV (CD26) in cultured peripheral blood T lymphocytes of oral cancer patients. J Oral Pathol Med 27:106–110
Annunziato F, Romagnani C, Romagnani S (2015) The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol 135:626–635
Robinson DS (2010) The role of the T cell in asthma. J Allergy Clin Immunol 126:1081–1091
Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, Huff R, Pilewski J, Holguin F, Kolls J, Wenzel S, Ray P, Ray A (2015) High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest 125:3037–3050
Nakajima H, Hirose K (2010) Role of IL-23 and Th17 cells in airway inflammation in asthma. Immune Netw 10:1–4
Zennaro D, Scala E, Pomponi D, Caprini E, Arcelli D, Gambineri E, Russo G, Mari A (2012) Proteomics plus genomics approaches in primary immunodeficiency: the case of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Clin Exp Immunol 167:120–128
Ohshima M, Futamura M, Kamachi Y, Ito K, Sakamoto T (2009) Allergic bronchopulmonary aspergillosis in a 2-year-old asthmatic boy with immune dysregulation, polyendocrinopathy, enteropathy, X-linked. Pediatr Pulmonol 44:297–299
Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, Thunberg S, Deniz G, Valenta R, Fiebig H, Kegel C, Disch R, Schmidt-Weber CB, Blaser K, Akdis CA (2004) Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med 199:1567–1575
Runyon RS, Cachola LM, Rajeshuni N, Hunter T, Garcia M, Ahn R, Lurmann F, Krasnow R, Jack LM, Miller RL, Swan GE, Kohli A, Jacobson AC, Nadeau KC (2012) Asthma discordance in twins is linked to epigenetic modifications of T cells. PLoS One 7:e48796
De Meester I, Vanham G, Kestens L, Vanhoof G, Bosmans E, Gigase P, Scharpé S (1994) Binding of adenosine deaminase to the lymphocyte surface via CD26. Eur J Immunol 24:566–570
Ishii T, Ohnuma K, Murakami A, Takasawa N, Kobayashi S, Dang NH, Schlossman SF, Morimoto C (2001) CD26-mediated signaling for T cell activation occurs in lipid rafts through its association with CD45RO. Proc Natl Acad Sci U S A 98:12138–12143
Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Yoshikawa N, Shimizu N, Iwata S, Tanaka H, Dang NH, Morimoto C (2004) CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc Natl Acad Sci U S A 101:14186–14191
Herrera C, Morimoto C, Blanco J, Mallol J, Arenzana F, Lluis C, Franco R (2001) Comodulation of CXCR4 and CD26 in human lymphocytes. J Biol Chem 276:19532–19539
Löster K, Zeilinger K, Schuppan D, Reutter W (1995) The cysteine-rich region of dipeptidyl peptidase IV (CD 26) is the collagen-binding site. Biochem Biophys Res Commun 217:341–348
Gonzalez-Gronow M, Grenett HE, Weber MR, Gawdi G, Pizzo SV (2001) Interaction of plasminogen with dipeptidyl peptidase IV initiates a signal transduction mechanism which regulates expression of matrix metalloproteinase-9 by prostate cancer cells. Biochem J 355:397–407
Davoodi J, Kelly J, Gendron NH, MacKenzie AE (2007) The Simpson-Golabi-Behmel syndrome causative glypican-3, binds to and inhibits the dipeptidyl peptidase activity of CD26. Proteomics 7:2300–2310
Piazza GA, Callanan HM, Mowery J, Hixson DC (1989) Evidence for a role of dipeptidyl peptidase IV in fibronectin-mediated interactions of hepatocytes with extracellular matrix. Biochem J 262:327–334
Cheng HC, Abdel-Ghany M, Pauli BU (2003) A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J Biol Chem 278:24600–24607
Komiya E, Ohnuma K, Yamazaki H, Hatano R, Iwata S, Okamoto T, Dang NH, Yamada T, Morimoto C (2014) CD26-mediated regulation of periostin expression contributes to migration and invasion of malignant pleural mesothelioma cells. Biochem Biophys Res Commun 447:609–615
Bobolea I, Barranco P, Del Pozo V, Romero D, Sanz V, López-Carrasco V, Canabal J, Villasante C, Quirce S (2015) Sputum periostin in patients with different severe asthma phenotypes. Allergy 70:540–546
Salgado FJ, Lojo J, Alonso-Lebrero JL, Lluis C, Franco R, Cordero OJ, Nogueira M (2003) A role for interleukin-12 in the regulation of T cell plasma membrane compartmentation. J Biol Chem 278:24849–24857
Saunders AE, Johnson P (2010) Modulation of immune cell signalling by the leukocyte common tyrosine phosphatase, CD45. Cell Signal 22:339–348
Hermiston ML, Xu Z, Weiss A (2003) CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol 21:107–137
Torimoto Y, Dang NH, Vivier E, Tanaka T, Schlossman SF, Morimoto C (1991) Coassociation of CD26 (dipeptidyl peptidase IV) with CD45 on the surface of human T lymphocytes. J Immunol 147:2514–2517
Hegen M, Kameoka J, Dong RP, Schlossman SF, Morimoto C (1997) Cross-linking of CD26 by antibody induces tyrosine phosphorylation and activation of mitogen-activated protein kinase. Immunology 90:257–264
Kähne T, Neubert K, Faust J, Ansorge S (1998) Early phosphorylation events induced by DPIV/CD26-specific inhibitors. Cell Immunol 189:60–66
Kobayashi S, Ohnuma K, Uchiyama M, Iino K, Iwata S, Dang NH, Morimoto C (2004) Association of CD26 with CD45RA outside lipid rafts attenuates cord blood T-cell activation. Blood 103:1002–1010
Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Iwata S, Hosono O, Kawasaki H, Tanaka H, Dang NH, Morimoto C (2005) CD26 mediates dissociation of Tollip and IRAK-1 from caveolin-1 and induces upregulation of CD86 on antigen-presenting cells. Mol Cell Biol 25:7743–7757
Ohnuma K, Uchiyama M, Yamochi T, Nishibashi K, Hosono O, Takahashi N, Kina S, Tanaka H, Lin X, Dang NH, Morimoto C (2007) Caveolin-1 triggers T-cell activation via CD26 in association with CARMA1. J Biol Chem 282:10117–10131
Ohnuma K, Yamochi T, Hosono O, Morimoto C (2005) CD26 T cells in the pathogenesis of asthma. Clin Exp Immunol 139:13–16
Hühn J, Ehrlich S, Fleischer B, von Bonin A (2000) Molecular analysis of CD26-mediated signal transduction in T cells. Immunol Lett 72:127–132
Vora KA, Porter G, Peng R, Cui Y, Pryor K, Eiermann G, Zaller DM (2009) Genetic ablation or pharmacological blockade of dipeptidyl peptidase IV does not impact T cell-dependent immune responses. BMC Immunol 10:19–29
Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C (1993) Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 261:466–469
Gracia E, Farré D, Cortés A, Ferrer-Costa C, Orozco M, Mallol J, Lluís C, Canela EI, McCormick PJ, Franco R, Fanelli F, Casadó V (2013) The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. FASEB J 27:1048–1061
Blackburn MR, Thompson LF (2012) Adenosine deaminase deficiency: unanticipated benefits from the study of a rare immunodeficiency. J Immunol 188:933–935
Ciruela F, Saura C, Canela EI, Mallol J, Lluis C, Franco R (1996) Adenosine deaminase affects ligand-induced signalling by interacting with cell surface adenosine receptors. FEBS Lett 380:219–223
Gracia E, Pérez-Capote K, Moreno E, Barkešová J, Mallol J, Lluís C, Franco R, Cortés A, Casadó V, Canela EI (2011) A2A adenosine receptor ligand binding and signalling is allosterically modulated by adenosine deaminase. Biochem J 435:701–709
Martín M, Huguet J, Centelles JJ, Franco R (1995) Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J Immunol 155:4630–4643
Pacheco R, Martinez-Navio JM, Lejeune M, Climent N, Oliva H, Gatell JM, Gallart T, Mallol J, Lluis C, Franco R (2005) CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc Natl Acad Sci U S A 102:9583–9588
Naval-Macabuhay I, Casanova V, Navarro G, García F, León A, Miralles L, Rovira C, Martinez-Navio JM, Gallart T, Mallol J, Gatell JM, Lluís C, Franco R, McCormick PJ, Climent N (2016) Adenosine deaminase regulates Treg expression in autologous T cell-dendritic cell cocultures from patients infected with HIV-1. J Leukoc Biol 99:349–359
Molon B, Gri G, Bettella M, Gómez-Moutón C, Lanzavecchia A, Martínez-A C, Mañes S, Viola A (2005) T cell costimulation by chemokine receptors. Nat Immunol 6:465–471
Tanaka T, Duke-Cohan JS, Kameoka J, Yaron A, Lee I, Schlossman SF, Morimoto C (1994) Enhancement of antigen-induced T-cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc Natl Acad Sci U S A 91:3082–3086
Schmitz T, Underwood R, Khiroya R, Bachovchin WW, Huber BT (1996) Potentiation of the immune response in HIV-1+ individuals. J Clin Invest 97:1545–1549
Yu DM, Slaitini L, Gysbers V, Riekhoff AG, Kähne T, Knott HM, De Meester I, Abbott CA, McCaughan GW, Gorrell MD (2011) Soluble CD26/dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand J Immunol 73:102–111
Ohnuma K, Munakata Y, Ishii T, Iwata S, Kobayashi S, Hosono O, Kawasaki H, Dang NH, Morimoto C (2001) Soluble CD26/dipeptidyl peptidase IV induces T cell proliferation through CD86 up-regulation on APCs. J Immunol 167:6745–6755
Ikushima H, Munakata Y, Ishii T, Iwata S, Terashima M, Tanaka H, Schlossman SF, Morimoto C (2000) Internalization of CD26 by mannose 6-phosphate/insulin-like growth factor II receptor contributes to T cell activation. Proc Natl Acad Sci U S A 97:8439–8444
Wronkowitz N, Görgens SW, Romacho T, Villalobos LA, Sánchez-Ferrer CF, Peiró C, Sell H, Eckel J (2014) Soluble DPP4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim Biophys Acta 1842:1613–1621
Lee DS, Lee ES, Alam MM, Jang JH, Lee HS, Oh H, Kim YC, Manzoor Z, Koh YS, Kang DG, Lee DH (2016) Soluble DPP-4 up-regulates toll-like receptors and augments inflammatory reactions, which are ameliorated by vildagliptin or mannose-6-phosphate. Metabolism 65:89–101
Proost P, Struyf S, Schols D, Opdenakker G, Sozzani S, Allavena P, Mantovani A, Augustyns K, Bal G, Haemers A, Lambeir AM, Scharpé S, Van Damme J, De Meester I (1999) Truncation of macrophage-derived chemokine by CD26/ dipeptidyl-peptidase IV beyond its predicted cleavage site affects chemotactic activity and CC chemokine receptor 4 interaction. J Biol Chem 274:3988–3993
Engel M, Hoffmann T, Manhart S, Heiser U, Chambre S, Huber R, Demuth HU, Bode W (2006) Rigidity and flexibility of dipeptidyl peptidase IV: crystal structures of and docking experiments with DPIV. J Mol Biol 354:768–783
Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpé S (1995) Proline motifs in peptides and their biological processing. FASEB J 9:736–744
Tanaka T, Kameoka J, Yaron A, Schlossman SF, Morimoto C (1993) The costimulatory activity of the CD26 antigen requires dipeptidyl peptidase IV enzymatic activity. Proc Natl Acad Sci U S A 90:4586–4590
Schön E, Mansfeld HW, Demuth HU, Barth A, Ansorge S (1985) The dipeptidyl peptidase IV, a membrane enzyme involved in the proliferation of T lymphocytes. Biomed Biochim Acta 44:K9–K15
Reinhold D, Bank U, Bühling F, Lendeckel U, Faust J, Neubert K, Ansorge S (1997) Inhibitors of dipeptidyl peptidase IV induce secretion of transforming growth factor-beta 1 in PWM-stimulated PBMC and T cells. Immunology 91:354–360
Reinhold D, Bank U, Bühling F, Täger M, Born I, Faust J, Neubert K, Ansorge S (1997) Inhibitors of dipeptidyl peptidase IV (DP IV, CD26) induces secretion of transforming growth factor-beta 1 (TGF-beta 1) in stimulated mouse splenocytes and thymocytes. Immunol Lett 58:29–35
Reinhold D, Hemmer B, Gran B, Born I, Faust J, Neubert K, McFarland HF, Martin R, Ansorge S (1998) Inhibitors of dipeptidyl peptidase IV/CD26 suppress activation of human MBP-specific CD4+ T cell clones. J Neuroimmunol 87:203–209
Tanaka S, Murakami T, Horikawa H, Sugiura M, Kawashima K, Sugita T (1997) Suppression of arthritis by the inhibitors of dipeptidyl peptidase IV. Int J Immunopharmacol 19:15–24
Steinbrecher A, Reinhold D, Quigley L, Gado A, Tresser N, Izikson L, Born I, Faust J, Neubert K, Martin R, Ansorge S, Brocke S (2000) Dipeptidyl peptidase IV in inflammatory CNS disease. Adv Exp Med Biol 477:145–153
Dai Y, Dai D, Wang X, Ding Z, Mehta JL (2014) DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc Drugs Ther 28:425–432
Wilson CN, Nadeem A, Spina D, Brown R, Page CP, Mustafa SJ (2009) Adenosine receptors and asthma. Handb Exp Pharmacol 329–362.
Polosa R, Blackburn MR (2009) Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol Sci 30:528–535
Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K, Iscove N, Koretzky G, Johnson P, Liu P, Rothstein DM, Penninger JM (2001) CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409:349–354
Stütz A, Graf P, Beinhauer B, Hammerschmid F, Neumann C, Woisetschläger M, Jung T (2005) CD45 isoform expression is associated with different susceptibilities of human naive and effector CD4+ T cells to respond to IL-4. Eur J Immunol 35:575–583
Mentlein R, Gallwitz B, Schmidt WE (1993) Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem 214:829–835
Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, McCaughan GW, Gorrell MD (2010) The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J 277:1126–1144
Boonacker E, Van Noorden CJ (2003) The multifunctional or moonlighting protein CD26/DPPIV. Eur J Cell Biol 82:53–73
Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, Messina-Graham S, Hangoc G, Farag S, Rohrabaugh SL, Ou X, Speth J, Pelus LM, Srour EF, Campbell TB (2012) Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat Med 18:1786–1796
Tasic T, Bäumer W, Schmiedl A, Schwichtenhövel F, Pabst R, Raap U, von Hörsten S, Stephan M (2011) Dipeptidyl peptidase IV (DPP4) deficiency increases Th1-driven allergic contact dermatitis. Clin Exp Allergy 41:1098–1107
Lankas GR, Leiting B, Roy RS, Eiermann GJ, Beconi MG, Biftu T, Chan CC, Edmondson S, Feeney WP, He H, Ippolito DE, Kim D, Lyons KA, Ok HO, Patel RA, Petrov AN, Pryor KA, Qian X, Reigle L, Woods A, Wu JK, Zaller D, Zhang X, Zhu L, Weber AE, Thornberry NA (2005) Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes 54:2988–2994
Preller V, Gerber A, Wrenger S, Togni M, Marguet D, Tadje J, Lendeckel U, Röcken C, Faust J, Neubert K, Schraven B, Martin R, Ansorge S, Brocke S, Reinhold D (2007) TGF-beta1-mediated control of central nervous system inflammation and autoimmunity through the inhibitory receptor CD26. J Immunol 178:4632–4640
Baticic L, Detel D, Kucic N, Buljevic S, Pugel EP, Varljen J (2011) Neuroimmunomodulative properties of dipeptidyl peptidase IV/CD26 in a TNBS-induced model of colitis in mice. J Cell Biochem 112:3322–3333
McGuinness C, Wesley UV (2008) Dipeptidyl peptidase IV (DPPIV), a candidate tumor suppressor gene in melanomas is silenced by promoter methylation. Front Biosci 13:2435–2443
Arscott WT, LaBauve AE, May V, Wesley UV (2009) Suppression of neuroblastoma growth by dipeptidyl peptidase IV: relevance of chemokine regulation and caspase activation. Oncogene 28:479–491
Oravecz T, Pall M, Roderiquez G, Gorrell MD, Ditto M, Nguyen NY, Boykins R, Unsworth E, Norcross MA (1997) Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med 186:1865–1872
Kufareva I, Salanga CL, Handel TM (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol Cell Biol 93:372–383
Wang J, Harada A, Matsushita S, Matsumi S, Zhang Y, Shioda T, Nagai Y, Matsushima K (1998) IL-4 and a glucocorticoid up-regulate CXCR4 expression on human CD4+ T lymphocytes and enhance HIV-1 replication. J Leukoc Biol 64:642–649
Gonzalo JA, Lloyd CM, Peled A, Delaney T, Coyle AJ, Gutierrez-Ramos JC (2000) Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1 alpha in the inflammatory component of allergic airway disease. J Immunol 165:499–508
Bai Z, Hayasaka H, Kobayashi M, Li W, Guo Z, Jang MH, Kondo A, Choi BI, Iwakura Y, Miyasaka M (2009) CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer’s patches. J Immunol 182:1287–1295
Werner L, Guzner-Gur H, Dotan I (2013) Involvement of CXCR4/CXCR7/CXCL12 Interactions in inflammatory bowel disease. Theranostics 3:40–46
Romagnani S (2002) Cytokines and chemoattractants in allergic inflammation. Mol Immunol 38:881–885
Islam SA, Luster AD (2012) T cell homing to epithelial barriers in allergic disease. Nat Med 18:705–715
Manns J, Rieder S, Escher S, Eilers B, Forssmann WG, Elsner J, Forssmann U (2007) The allergy-associated chemokine receptors CCR3 and CCR5 can be inactivated by the modified chemokine NNY-CCL11. Allergy 62:17–24
Sehmi R, Dorman S, Baatjes A, Watson R, Foley R, Ying S, Robinson DS, Kay AB, O’Byrne PM, Denburg JA (2003) Allergen-induced fluctuation in CC chemokine receptor 3 expression on bone marrow CD34+ cells from asthmatic subjects: significance for mobilization of haemopoietic progenitor cells in allergic inflammation. Immunology 109:536–546
Lambeir AM, Proost P, Durinx C, Bal G, Senten K, Augustyns K, Scharpé S, Van Damme J, De Meester I (2001) Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase IV reveals a striking selectivity within the chemokine family. J Biol Chem 276:29839–29845
Struyf S, Proost P, Schols D, De Clercq E, Opdenakker G, Lenaerts JP, Detheux M, Parmentier M, De Meester I, Scharpé S, Van Damme J (1999) CD26/dipeptidyl-peptidase IV down-regulates the eosinophil chemotactic potency, but not the anti-HIV activity of human eotaxin by affecting its interaction with CC chemokine receptor 3. J Immunol 162:4903–4909
Forssmann U, Stoetzer C, Stephan M, Kruschinski C, Skripuletz T, Schade J, Schmiedl A, Pabst R, Wagner L, Hoffmann T, Kehlen A, Escher SE, Forssmann WG, Elsner J, von Hörsten S (2008) Inhibition of CD26/dipeptidyl peptidase IV enhances CCL11/eotaxin-mediated recruitment of eosinophils in vivo. J Immunol 181:1120–1127
Savino B, Borroni EM, Torres NM, Proost P, Struyf S, Mortier A, Mantovani A, Locati M, Bonecchi R (2009) Recognition versus adaptive up-regulation and degradation of CC chemokines by the chemokine decoy receptor D6 are determined by their N-terminal sequence. J Biol Chem 284:26207–26215
Pawig L, Klasen C, Weber C, Bernhagen J, Noels H (2015) Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol 6:429
Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ (2002) AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 160:1353–1360
Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA (1996) A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med 184:1101–1109
Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE (2000) Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol 165:6590–6598
Ohtsuki T, Hosono O, Kobayashi H, Munakata Y, Souta A, Shioda T, Morimoto C (1998) Negative regulation of the anti-human immunodeficiency virus and chemotactic activity of human stromal cell-derived factor 1alpha by CD26/dipeptidyl peptidase IV. FEBS Lett 431:236–240
Proost P, Struyf S, Schols D, Durinx C, Wuyts A, Lenaerts JP, De Clercq E, De Meester I, Van Damme J (1998) Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1alpha. FEBS Lett 432:73–76
De La Luz SM, Yang F, Narazaki M, Salvucci O, Davis D, Yarchoan R, Zhang HH, Fales H, Tosato G (2004) Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood 103:2452–2459
Christopherson KW 2nd, Hangoc G, Broxmeyer HE (2002) Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. J Immunol 169:7000–7008
Iwata S, Yamaguchi N, Munakata Y, Ikushima H, Lee JF, Hosono O, Schlossman SF, Morimoto C (1999) CD26/dipeptidyl peptidase IV differentially regulates the chemotaxis of T cells and monocytes toward RANTES: possible mechanism for the switch from innate to acquired immune response. Int Immunol 11:417–426
Ohtsuki T, Tsuda H, Morimoto C (2000) Good or evil: CD26 and HIV infection. J Dermatol Sci 22:152–160
Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR (1997) The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A 94:1925–1930
Franitza S, Kollet O, Brill A, Vaday GG, Petit I, Lapidot T, Alon R, Lider O (2002) TGF-beta1 enhances SDF-1alpha-induced chemotaxis and homing of naive T cells by up-regulating CXCR4 expression and downstream cytoskeletal effector molecules. Eur J Immunol 32:193–202
Post S, Smits AM, van den Broek AJ, Sluijter JP, Hoefer IE, Janssen BJ, Snijder RJ, Mager JJ, Pasterkamp G, Mummery CL, Doevendans PA, Goumans MJ (2010) Impaired recruitment of HHT-1 mononuclear cells to the ischaemic heart is due to an altered CXCR4/CD26 balance. Cardiovasc Res 85:494–502
Post S, van den Broek AJ, Rensing BJ, Pasterkamp G, Goumans MJ, Doevendans PA (2012) Reduced CD26 expression is associated with improved cardiac function after acute myocardial infarction: insights from the REPERATOR study. J Mol Cell Cardiol 53:899–905
Havre PA, Abe M, Urasaki Y, Ohnuma K, Morimoto C, Dang NH (2009) CD26 expression on T cell lines increases SDF-1-alpha-mediated invasion. Br J Cancer 101:983–991
Fernandis AZ, Cherla RP, Ganju RK (2003) Differential regulation of CXCR4-mediated T-cell chemotaxis and mitogen-activated protein kinase activation by the membrane tyrosine phosphatase, CD45. J Biol Chem 278:9536–9543
Mantovani A, Gray PA, Van Damme J, Sozzani S (2000) Macrophage-derived chemokine (MDC). J Leukoc Biol 68:400–404
Yamashita U, Kuroda E (2002) Regulation of macrophage-derived chemokine (MDC, CCL22) production. Crit Rev Immunol 22:105–114
Yanai M, Sato K, Aoki N, Takiyama Y, Oikawa K, Kobayashi H, Kimura S, Harabuchi Y, Tateno M (2007) The role of CCL22/macrophage-derived chemokine in allergic rhinitis. Clin Immunol 125:291–298
Kim CH, Rott L, Kunkel EJ, Genovese MC, Andrew DP, Wu L, Butcher EC (2001) Rules of chemokine receptor association with T cell polarization in vivo. J Clin Invest 108:1331–1339
Appay V, Rowland-Jones SL (2001) RANTES: a versatile and controversial chemokine. Trends Immunol 22:83–87
Lim JK, Burns JM, Lu W, DeVico AL (2005) Multiple pathways of amino terminal processing produce two truncated variants of RANTES/CCL5. J Leukoc Biol 78:442–452
Proost P, Menten P, Struyf S, Schutyser E, De Meester I, Van Damme J (2000) Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood 96:1674–1680
Nakao M, Nomiyama H, Shimada K (1990) Structures of human genes coding for cytokine LD78 and their expression. Mol Cell Biol 10:3646–3658
Menten P, Struyf S, Schutyser E, Wuyts A, De Clercq E, Schols D, Proost P, Van Damme J (1999) The LD78beta isoform of MIP-1alpha is the most potent CCR5 agonist and HIV-1-inhibiting chemokine. J Clin Invest 104:R1–R5
Struyf S, Menten P, Lenaerts JP, Put W, D’Haese A, De Clercq E, Schols D, Proost P, Van Damme J (2001) Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein-1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti-HIV-1 activity and chemotactic potencies for neutrophils and eosinophils. Eur J Immunol 31:2170–2178
Ludwig A, Schiemann F, Mentlein R, Lindner B, Brandt E (2002) Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I-TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J Leukoc Biol 72:183–191
Proost P, Mortier A, Loos T, Vandercappellen J, Gouwy M, Ronsse I, Schutyser E, Put W, Parmentier M, Struyf S, Van Damme J (2007) Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood 110:37–44
Casrouge A, Bisiaux A, Stephen L, Schmolz M, Mapes J, Pfister C, Pol S, Mallet V, Albert ML (2012) Discrimination of agonist and antagonist forms of CXCL10 in biological samples. Clin Exp Immunol 167:137–148
Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646
Pennino D, Bhavsar PK, Effner R, Avitabile S, Venn P, Quaranta M, Marzaioli V, Cifuentes L, Durham SR, Cavani A, Eyerich K, Chung KF, Schmidt-Weber CB, Eyerich S (2013) IL-22 suppresses IFN-γ-mediated lung inflammation in asthmatic patients. J Allergy Clin Immunol 131:562–570
Kruschinski C, Skripuletz T, Bedoui S, Tschernig T, Pabst R, Nassenstein C, Braun A, von Hörsten S (2005) CD26 (dipeptidyl-peptidase IV)-dependent recruitment of T cells in a rat asthma model. Clin Exp Immunol 139:17–24
Tsuji E, Misumi Y, Fujiwara T, Takami N, Ogata S, Ikehara Y (1992) An active-site mutation (Gly633 → Arg) of dipeptidyl peptidase IV causes its retention and rapid degradation in the endoplasmic reticulum. Biochemistry 31:11921–11927
Schmiedl A, Krainski J, Schwichtenhövel F, Schade J, Klemann C, Raber KA, Zscheppang K, Beekmann T, Acevedo C, Glaab T, Wedekind D, Pabst R, von Hörsten S, Stephan M (2010) Reduced airway inflammation in CD26/DPP4-deficient F344 rats is associated with altered recruitment patterns of regulatory T cells and expression of pulmonary surfactant proteins. Clin Exp Allergy 40:1794–1808
Schade J, Schmiedl A, Stephan M, Pabst R, von Hörsten S (2010) Transferred T cells preferentially adhere in the BALT of CD26-deficient recipient lungs during asthma. Immunobiology 215:321–331
Narducci MG, Scala E, Bresin A, Caprini E, Picchio MC, Remotti D, Ragone G, Nasorri F, Frontani M, Arcelli D, Volinia S, Lombardo GA, Baliva G, Napolitano M, Russo G (2006) Skin homing of Sézary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood 107:1108–1115
Hoshino M, Aoike N, Takahashi M, Nakamura Y, Nakagawa T (2003) Increased immunoreactivity of stromal cell-derived factor-1 and angiogenesis in asthma. Eur Respir J 21:804–809
Tasic T, Stephan M, von Hörsten S, Pabst R, Schmiedl A (2014) Differential OVA-induced pulmonary inflammation and unspecific reaction in Dark Agouti (DA) rats contingent on CD26/DPPIV deficiency. Immunobiology 219:888–900
Conarello SL, Li Z, Ronan J, Roy RS, Zhu L, Jiang G, Liu F, Woods J, Zycband E, Moller DE, Thornberry NA, Zhang BB (2003) Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc Natl Acad Sci U S A 100:6825–6830
Yan S, Marguet D, Dobers J, Reutter W, Fan H (2003) Deficiency of CD26 results in a change of cytokine and immunoglobulin secretion after stimulation by pokeweed mitogen. Eur J Immunol 33:1519–1527
Rogala B, Glück J, Mazur B (2002) Do the molecules CD26 and lymphocytes activation gene-3 differentiate between type 1 and 2 T cell response? J Investig Allergol Clin Immunol 12:198–203
Matsuno O, Miyazaki E, Nureki S, Ueno T, Ando M, Kumamoto T (2007) Soluble CD26 is inversely associated with disease severity in patients with chronic eosinophilic Pneumonia. Biomark Insights 1:201–204
Katoh N, Hirano S, Suehiro M, Ikenaga K, Yamashita T, Sugawara N, Yasuno H (2000) Soluble CD30 is more relevant to disease activity of atopic dermatitis than soluble CD26. Clin Exp Immunol 121:187–192
Leiria LO, Martins MA, Saad MJ (2015) Obesity and asthma: beyond T(H)2 inflammation. Metabolism 64:172–181
Kim SH, Sutherland ER, Gelfand EW (2014) Is there a link between obesity and asthma? Allergy, Asthma Immunol Res 6:189–195
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I (2013) Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 369:1317–1326
Raschi E, Poluzzi E, Koci A, Antonazzo IC, Marchesini G, De Ponti F (2016) Dipeptidyl peptidase-4 inhibitors and heart failure: analysis of spontaneous reports submitted to the FDA Adverse Event Reporting System. Nutr Metab Cardiovasc Dis 26:380–386
Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR (2015) Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 373:232–242
Willemen MJ, Mantel-Teeuwisse AK, Straus SM, Meyboom RH, Egberts TC, Leufkens HG (2011) Use of dipeptidyl peptidase-4 inhibitors and the reporting of infections: a disproportionality analysis in the World Health Organization VigiBase. Diabetes Care 34:369–374
Amori RE, Lau J, Pittas AG (2007) Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA 298:194–206
Monami M, Iacomelli I, Marchionni N, Mannucci E (2010) Dipeptydil peptidase-4 inhibitors in type 2 diabetes: a meta-analysis of randomized clinical trials. Nutr Metab Cardiovasc Dis 20:224–235
Béné J, Jacobsoone A, Coupe P, Auffret M, Babai S, Hillaire-Buys D, Jean-Pastor MJ, Vonarx M, Vermersch A, Tronquoy AF, Gautier S (2015) Bullous pemphigoid induced by vildagliptin: a report of three cases. Fundam Clin Pharmacol 29:112–114
El-Hashim AZ, Amine SA (2005) The role of substance P and bradykinin in the cough reflex and bronchoconstriction in guinea-pigs. Eur J Pharmacol 513:125–133
Ramalho R, Almeida J, Beltrão M, Pirraco A, Costa R, Sokhatska O, Guardão L, Palmares C, Guimarães JT, Delgado L, Moreira A, Soares R (2012) Substance P antagonist improves both obesity and asthma in a mouse model. Allergy 68:48–54
Sromova L, Busek P, Posova H, Potockova J, Skrha P, Andel M, Sedo A (2016) The effect of dipeptidyl peptidase-IV inhibition on circulating T cell subpopulations in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 118:183–192
Lee YS, Jun HS (2016) Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm 2016:3094642
Brightling CE, Chanez P, Leigh R, O’Byrne PM, Korn S, She D, May RD, Streicher K, Ranade K, Piper E (2015) Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 3:692–701
Shiobara T, Chibana K, Watanabe T, Arai R, Horigane Y, Nakamura Y, Hayashi Y, Shimizu Y, Takemasa A, Ishii Y (2016) Dipeptidyl peptidase-4 is highly expressed in bronchial epithelial cells of untreated asthma and it increases cell proliferation along with fibronectin production in airway constitutive cells. Respir Res 17:28
Matteucci E, Giampietro O (2009) Dipeptidyl peptidase-4 (CD26): knowing the function before inhibiting the enzyme. Curr Med Chem 16:2943–2951
Vanderheyden M, Bartunek J, Goethals M, Verstreken S, Lambeir AM, De Meester I, Scharpé S (2009) Dipeptidyl-peptidase IV and B-type natriuretic peptide. From bench to bedside. Clin Chem Lab Med 47:248–252
Zhu L, Tamvakopoulos C, Xie D, Dragovic J, Shen X, Fenyk-Melody JE, Schmidt K, Bagchi A, Griffin PR, Thornberry NA, Sinha Roy R (2003) The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides: in vivo metabolism of pituitary adenylate cyclase activating polypeptide-(1–38). J Biol Chem 278:22418–22423
Acknowledgments
This project was funded by Instituto de Salud Carlos III, Ministerio de Economía y Competitividad (PI13/02046) and Sociedad Española de Neumología y Cirugía Torácica, SEPAR (121/2012).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nieto-Fontarigo, J.J., González-Barcala, F.J., San José, E. et al. CD26 and Asthma: a Comprehensive Review. Clinic Rev Allerg Immunol 56, 139–160 (2019). https://doi.org/10.1007/s12016-016-8578-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8578-z