
Abstract
Purpose of Review
Giant cell arteritis (GCA), a medium and large vessel vasculitis occurring in the aged, remains a formidable disease, capable of taking both vision and life, through a multitude of vascular complications. Our understanding of the spectrum of its manifestations has grown over the years, to include limb claudication, aortitis, and cardiac disease, in addition to the more classic visual complications resulting from of ischemia to branches of the external and internal carotid arteries. While a clinical presentation of headache, jaw claudication, scalp tenderness, fever and other systemic symptoms and serum markers are together highly suggestive of the disease, diagnosis can be challenging in those cases in which classic symptoms are lacking. The purpose of this review is to update the reader on advances in the diagnosis and treatment of giant cell arteritis and to review our evolving understanding of the immunological mechanism underlying the disease, which have helped guide our search for novel therapies.
Recent Findings
There is increasing evidence supporting the use of Doppler ultrasound, dedicated post-contrast T1-weighted spin echo MRI of the scalp arteries and PET scan, which can together improve our diagnostic accuracy in cases in which temporal artery biopsy is either inconclusive or not feasible. Advances in our understanding of the immunological cascades underlying the disease have helped guide our search for steroid-sparing treatments for the GCA, the most important of which has been the IL-6 receptor antibody inhibitor tocilizumab, which has been shown to reduce cumulative steroid dose in a large multicenter, placebo-controlled prospective study. Other biologic agents, such as abatacept and ustekinumab have shown promise in smaller studies.
Summary
GCA is no longer a disease whose diagnosis is based exclusively on temporal artery biopsy and whose complications are prevented solely with the use of corticosteroids. Modern vascular imaging techniques and targeted immunologic therapies are heralding a new era for the disease, in which practitioners will hopefully be able to diagnosis it with greater accuracy and treat it with less ischemic complications and iatrogenic side effects.
Similar content being viewed by others


Introduction
Temporal arteritis, also known as giant cell arteritis (GCA), in recognition of its typical but not ubiquitous histological finding, is a granulomatous vasculitis of medium and large vessels that mostly affects the elderly. While our understanding of the pathophysiology underlying the disease has grown over the years, with some lines of evidence pointing to an infectious etiology, a clear-cut environmental cause remains elusive. Its chief vascular targets are extracranial branches of the external and internal carotid arteries, the former resulting in headache, scalp tenderness and jaw claudication, and the latter resulting in potentially blinding ischemic injury to the optic nerve or retina. In fact, at least 9% of patients experience severe, irreversible vision loss from the disease [1], a complication that is more likely if high dose corticosteroids are not started in a timely fashion. Less common but life-threatening complications include myocardial infarction, cerebral infarction and aortic aneurysms. Modern imaging techniques, including Doppler ultrasound of the temporal artery, specialized MRI sequences and FDG-PET scans, have gained acceptance as methods of diagnosis, either in addition to, or in some cases, instead of, temporal artery biopsy. Management of this difficult disease has evolved over the years, to now include tocilizumab, an interleukin-6 (IL-6) inhibitor that may allow for a quicker reduction in steroid doses, although treatment typically must be continued for a year or more. With its potential for blindness and even death, GCA should be considered in any case of unexplained headache in patients over 50, and no time should be wasted in reaching the diagnosis so that treatment can be started quickly, preventing vision loss or worse.
Historical perspective
Horton named the disease temporal arteritis in a description of two cases in 1932 (earning the eponym “Horton’s disease”) and later went on to conclude that it was an autoimmune disease, after performing temporal artery injections into patients with quiescent disease, producing relapses [2]. However, the disease had been reported first by Hutchinson in 1890, who described what he called “thrombotic arteritis of the aged,” in an 80-year-old man who presented with swollen red streaks on his temple that became thrombosed “cords” [3]. One may wonder if GCA existed prior to the 1800s since no prior descriptions have been found in the medical literature, but stigmata of the disease have been noticed in several paintings from as early as the 1400s [4].
Pathophysiology
An understanding of the immunologic and vascular basis of GCA is helpful before embarking on any discussion of its therapeutics. GCA pathophysiology develops along two CD4 cell axes culminating in a systemic inflammatory response and vascular occlusion. The activation of dendritic cells within the adventitia of blood vessels is likely the result of some environmental or infectious stimulus, via activation of their pattern recognition receptors such as the toll like receptor (TLR) and results in the release of chemokines (CCL18-21) that recruit naive CD4+ helper T cells into the layers of the arterial wall where interleukins (IL) released by the dendritic cell activate them [5]. Under the influence of IL-6, some CD4+ cells differentiate into Th17 cells which produce IL-17 [6]. Il-6 may at the same time lead to a reduction in regulatory T cells (Treg), thus reducing one protective arm against the immunological cascade.
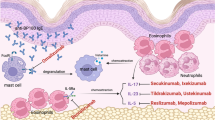
Other CD4+ cells, triggered by IL-12 and IL-18, differentiate into Th1 cells which release interferon gamma (INF-γ), which in turn induces vascular smooth muscle cells (VSM) to release cytokines that recruit monocytes which either differentiate into macrophages (under the influence of IFN-γ) or fuse to form the eponymous multinucleated giant cells [7]. The macrophages produce more IL-6 as well as TNF-α, which leads to many of the systemic symptoms of GCA such as fever and fatigue. These IFN-γ activated macrophages drive the vascular remodeling by releasing reactive oxygen species that peroxides phospholipids in cellular membranes and metalloproteinase-2 (MMP-2) which, along with MMP-9 released by VSM, destroy cellular matrix proteins such as elastin, resulting in the liquidation of the media and internal elastic lamina. Other VSM-released cytokines recruit more Th1 cells, leading to a positive feedback loop perpetuating the response. Still other cytokines recruit CD8+ T cells which infiltrate the arteriole wall and release cytotoxic perforin and granzymes. Finally, macrophages, injured VSM and giant cells all release platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which are responsible for hyperplasia of the intima and neoangiogenesis respectively. Intimal hyperplasia is driven by PDGF-induced VSM migration to the intima; a role is therefore suggested for the PDGF-blocking drug imatinib in the disease [8]. Both the intimal hyperplasia and neoangiogenesis ultimately lead to vascular occlusion and the ischemic complications of GCA. While the complex biochemical contributors to the pathogenesis of GCA present numerous potential targets for steroid-sparing therapy, a correlation between IL-6 levels and disease activity in GCA [9] has supported the evaluation of IL-blockade specifically in the treatment of the disease. See Fig. 1.

Pathogenesis of giant cell arteritis. Unknown environmental stimuli, possibly infectious, activate immature dendritic cells within the adventitia of blood vessels through stimulation of receptors such as the toll like receptor (TLR), leading to the release of chemokines (CCL18–21) that recruit naive CD4+ helper T cells. These T cells, under the influence of interleukin-6 (IL-6), differentiate into Th17 cells which produce IL-17, while others, triggered by IL-12, differentiate into Th1 cells which release interferon gamma (INF-γ). Il-6 and IL-17 may at the same time lead to a reduction in regulatory T cells (Treg). INF-γ causes vascular smooth muscle cells (VSM) to release cytokines that recruit monocytes which are transformed into either macrophages or multinucleated giant cells under the influence of INF-γ. Macrophages release reactive oxygen species that peroxides phospholipids in cellular membranes and matrix metalloproteinase-2 (MMP-2) which, along with MMP-9 released by VSM, destroy cellular matrix proteins such as elastin, resulting in the destruction of the media. CXCL10–11 released by VSM cells recruit CD8+ T cells which release cytotoxic perforin. Macrophages, injured VSM, and giant cells all release platelet-derived growth factor (PDGF) which leads to intimal hyperplasia and associated luminal stenosis, which in turn can lead to luminal thrombosis. Vascular endothelial growth factor (VEGF) released by giant cells leads to neoangiogenesis. TLR, troll-like receptor. CD4, undifferentiated CD4+ helper T cell. INF-γ, interferon gamma. VSM, vascular smooth muscle cells. O-, reactive oxygen species. NO, nitric oxide. Treg, regulatory T cell. MMP, matrix metalloproteinase. CD8, CD8+ killer T cells. PDGF, platelet-derived growth factor. VEGF, vascular endothelial growth factor. NEO, neoangiogenesis. IEL, internal elastic lamina.
Infectious hypothesis
While the etiology of GCA remains unknown, it has long been speculated that an infectious trigger is the inciting event, especially as its target population of elderly patients tends to be more susceptible to infections and less prone to primary autoimmune disease. In 2015, Gilden and colleagues speculated that varicella-zoster virus (VZV) was the environmental trigger of the immunologic response of GCA, based on similar pathological findings (e.g., granulomas, multinucleated giant cells) in both diseases, and prior case reports of VZV vasculitis mimicking GCA [10]. Their group stained thinly sliced temporal artery (TA) sections with mouse monoclonal anti VZV IgG1 antibody in 82 patients with histopathologically confirmed GCA [11•]. VZV antigen was identified in 74% of positive temporal artery biopsies (TABs) compared to 8% of controls and VZV antigen was found in skip lesions, adjacent to areas of GCA pathology. However, it is notable that the control vessels came from cadaveric temporal arteries, as opposed to live patients, which may have contributed to the difference. Furthermore, non-specific reactivity of VZV antibodies in a broad range of myocyte types and arteries, in diverse clinical settings, suggests that positivity reflects shared epitopes with VZV, rather than actual infection [12], and other more recent studies have failed to replicate Gilden’s findings [13, 14]. Other infectious hypotheses—herpes simplex virus, Epstein–Barr virus [15], parvovirus B19, chlamydia pneumonia, and mycoplasma pneumonia—have also emerged, but evidence remains scarce [16]. Whether or not an infectious agent plays a direct role in the pathophysiology of GCA, it is feasible that certain infections might trigger the inflammation of GCA, as suggested by higher rate antecedent infections in GCA patients compared with age-matched controls, especially upper respiratory infections [17].
Epidemiology
Giant cell arteritis is the most common systemic vasculitis affecting large and medium vessels, with advanced age being the most significant risk factor. While the disease steadily increases after age 50, its peak incidence is observed between 70 and 79 years of age [18]. A recent population based study in Ontario, Canada demonstrated a relatively stable incidence with 25 new cases per 100,000 people > 50 years of age [19], somewhat higher than the incidence in Olmsted County over a 50 year period (18 cases per 100,000 people > 50) reported in 2004 [20]. The highest incidence is found amongst individuals of Scandinavian descent, specifically in Norway, where the mean annual incidence was 32.8 per 100,000 inhabitants over age 50 years and 29.1 for biopsy proven GCA [21]. A much lower annual incidence has been reported in southern European countries and the Mediterranean region [22]. A recent study comparing patients with GCA in Italy to those in Olmsted county found that the Italian patients had a longer duration of symptoms prior to diagnosis, were younger, more likely to have cranial symptoms, permanent vision loss and systemic symptoms, and had higher serological markers as well [23]. A seasonal peak in May and June has been reported in a recent study in Jerusalem [24].
Risk factors for GCA other than age may include raised diastolic blood pressure and a history of smoking [25•]. However, a recent study found a negative association between fasting blood glucose levels and risk of GCA, suggesting that lower levels of glucose metabolites like pyruvate might lead to reduced expression of programmed death ligand 1 (PD-L1) on macrophages, predisposing to T cell activation and exacerbation of chronic vascular inflammation in GCA [26]. A recent study in Norway did not find worsened mortality in patients with GCA, although mortality in male GCA patients was worse than females [27]. However, a French study did find increased mortality in GCA patients, especially from vascular disease [28].
Manifestations
The most common premonitory symptom of GCA is new-onset headache in a patient over 50 years of age. Systemic symptoms including fever of unknown origin, anorexia and weight loss, and symptoms of polymyalgia rheumatica—shoulder and pelvic girdle stiffness and pain - may herald the disease, and warrant investigation. Owing to the intracranial and extracranial vascular inflammation of the aorta and its distal branches, the insidious and protean manifestations of GCA often make diagnosis difficult. Vision loss results from either arteritic anterior ischemic optic neuropathy (AAION) or central retinal artery occlusion (CRAO), either of which may be preceded by transient monocular vision loss, resulting from ischemia of the optic nerve, retina or choroid, occurring up to 8.5 days prior to vision loss [29]. Diplopia, resulting from microvascular ischemia to cranial nerves III, IV or VI, occurs in up to 6% of patients [30]. In one retrospective study of patients diagnosed with vision loss from GCA, amaurosis fugax was the initial presenting sign in 18% of cases and, of the patients with vision loss or blurred vision at the time of diagnosis, 44% experienced transient visual symptoms prior to treatment [1]. A work up for GCA, beginning with stat CBC, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), should therefore be part of the work up for any patient with amaurosis fugax over age 50, even if they do not have systemic symptoms. It should be noted that both ESR and CRP increase with age, and values should be assessed based on age-corrected reference ranges. Care should be made to note the units of CRP, which is sometimes reported in mg/L and sometimes in mg/dL.
Concomitant ischemic stroke occurs in up to 16% of patients with vision loss from GCA. Involvement of the intracranial branches of the aorta may result in internal carotid artery and vertebrobasilar territory symptoms and may be the initial presentation of the disease. Patients presenting with CRAO and AAION associated with GCA should therefore be evaluated with MR angiogram (MRA) of the head and neck.
Although the neurologist typically sees intracranial complications of GCA, extracranial vascular presentations may also occur. Aortitis is a widely recognized presentation of GCA and coronary artery involvement may lead to myocardial infarction [31]. Rare complications include pericardial effusion [32], tongue necrosis [33] and interstitial lung disease [34]. See Fig. 2 for a summary of GCA manifestations.

Manifestations of giant cell arteritis. Common manifestations include vision loss from anterior ischemic optic neuropathy, posterior ischemic optic neuropathy, central retinal artery occlusion, jaw claudication headache and scalp tenderness and systemic symptoms such as fever, fatigue, arthralgias, and myalgias. Less common symptoms include scalp or tongue necrosis, stroke, hearing loss, limb claudication, myocardial infarction, aortitis, and pulmonary fibrosis.
Orbital inflammation in giant cell arteritis
Giant cell arteritis may rarely manifest as orbital inflammation [35], with associated diplopia due to restriction of extraocular muscles, chemosis, proptosis and enhancement of the perineural optic sheaths [36]. In most cases, other symptoms and signs of the disease are present, although the radiological appearance does not distinguish it from other more common causes of orbital inflammation. Elderly patients presenting with orbital inflammation should be considered for GCA before idiopathic orbital inflammation is diagnosed, and temporal artery biopsy should be pursued if serum markers of inflammation are elevated. In some cases, a biopsy of orbital soft tissue has been confirmatory, demonstrating giant cells, fibrosis and necrosis [37].
Occult disease
Hayreh and colleagues reviewed 85 cases of biopsy-confirmed GCA in which there was some form of ocular involvement and found that 18 cases (21%) presented without any symptoms of systemic disease such as headache or fatigue [38]. While ESR and CRP were still elevated in this group, they were significantly lower than those with systemic symptoms. Rarely, serum marker of inflammation have been normal, which along with the lack of systemic symptoms can make for a challenging diagnosis [39]. The incidence of ocular involvement in occult GCA patients was A-AION in 94%, CRAO in 11.1%, amaurosis in 33.3% and diplopia in 11.1%.
See Fig. 3 for a classic case of GCA.

A case of giant cell arteritis. A 92-year-old woman complained of headache, jaw claudication, and vision loss. a Humphrey visual fields showed a superior altitudinal visual field loss in her right eye. b Funduscopy revealed a focal swelling inferiorly in the right optic disc that was also pale, i.e., pallid edema. Erythrocyte sedimentation rate (ESR) was 105 mm/h, C-reactive protein (CRP) was 3.6 dG/L. A temporal artery biopsy showed a mixed inflammatory infiltrate including giant cells within the arterial wall, consistent with GCA. c Elastin stain at low power demonstrate disruption of the internal elastic lamina (IEL) as well as a mixed infiltrate (MI) and intimal hyperplasia (IH). d Low power hematoxylin and eosin (H&E) demonstrates intimal hyperplasia (IH) and a mixed infiltrate (MI). e High power H&E demonstrates disruption of the IEL and a mixed infiltrate (MI). f High power H&E shows giant cells. (GC).
Diagnostic evaluation
Aside from the protean manifestations of GCA, the dilemma in patients with symptomatology concerning for GCA is overdiagnosis and exposure to high dose glucocorticoids in this vulnerable population, which has its own complications—hypertension, diabetes, osteopenia, and fractures. Regardless, the cost of a missed diagnosis may be even more catastrophic, potentially resulting in vision loss or even death from stroke or myocardial infarction. Since 1990, the mainstay of GCA diagnosis was predicated on fulfilling 3/5 of the America College of Rheumatology’s criteria, namely: age of onset ≥ 50 years, new onset of localized headache, temporal artery tenderness or decreased pulse, elevated ESR ≥ 50 mm/h and predominance of mononuclear cell infiltrates or a granulomatous inflammation with multinucleate giant cells on TAB [40]. Thus, while elevated inflammatory markers are important, their absence does not rule out the disease. The sensitivities of ESR > 50 mm/h, CRP > 20 mg/L, and platelets > 300 × 109/L were 65.5%, 66.9%, and 71.2% respectively, while specificities were 57.3%, 67.9%, and 71.2% respectively in one study of 139 patients, when compared against physician-determined diagnosis [41]. In the same analysis, if just ESR and CRP were positive, with normal platelets, the odds ratio of GCA was 4.0, which increased to 10.7 if platelets were high as well. Revised ACR criteria have been proposed, in which criteria are weighted with either a point score of 1 or 2, and a total of 3/11 points is considered positive, provided one of the criteria is either new-onset localized headache, sudden visual disturbance, polymyalgia rheumatica, jaw claudication, or abnormal temporal artery [42].
Temporal artery biopsy
Temporal artery biopsy remains an important element in the diagnosis of GCA, but its limitations included a delay in results, its invasive nature and imperfect sensitivity. While it is known that skip lesions may result in false negative results, a Bayesian analysis using multiple studies where bilateral temporal arteries were biopsied found a sensitivity of 87.1% [43]. It is an accepted notion that TAB should not be delayed beyond 14 days of corticosteroid treatment to avoid false negative results. However, a large study comparing results in patients treated with steroids for variable periods of time prior to biopsy found that 14 days or more of steroid therapy did not affect the positivity rate, although the pathological findings were more likely to be atypical [44].
These results are buttressed by a recent review of 3057 biopsies from a Veterans Health Administration National Database Cohort which found that positive TAB rates correlated with a postfixation biopsy length > 3.0 cm and bilateral biopsy in one sitting, but not with prednisone administration even beyond 42 days [45•]. A recent audit of pathologists found that there was complete agreement on the diagnosis in only 1/9 circulated images, highlighting the need for a standardized pathology TAB reporting template [46], although we feel that agreement between pathologists would likely be higher if they were allowed to review complete specimens.
To address the limitations of TAB, timely diagnostic imaging modalities have therefore been increasing employed to expediate treatment and prevent overdiagnosis and treatment complications.
Ultrasound
High-frequency color Doppler ultrasound (CDUS) has emerged as the imaging modality of choice in many institutions owing to advantage as a quick, cheap and non-invasive procedure that can detect vascular inflammation of the temporal and axillary arteries to support GCA diagnosis. The characteristic finding of GCA on US is the halo sign, which was defined by the OMERACT (Outcome Measures in Rheumatology group) as a homogeneous, hypoechoic wall thickening, best observed on the luminal side, which is visible on both longitudinal and transverse imaging [47]. The TABUL (Temporal Artery Biopsy vs ULtrasound in Diagnosis of GCA) study followed GCA suspects who underwent CDUS of both the TA and the axillary artery, during the first week of steroid treatment prospectively, and demonstrated that the halo sign, which reflects vessel wall edema and intimal thickness, had a similar sensitivity compared to TAB (93% vs. 91%), although it was less specific (77% vs. 81%) [48•]. In fact, in a recent study of 305 patients undergoing TA CDUS, 14 (4.6%) were felt to be false positive, based on the clinical course of the patients, with alternative diagnoses including neurosyphilis, lymphoma, granulomatosis with polyangiitis (GPA), PMR, and atherosclerosis [49]. Reasons for false positive results include arterial wall deposition of non-arteritic material (atherosclerosis and multiple myeloma), cellular infiltrates (lymphoma) and technical error. A meta-analysis looking at the results of 20 studies evaluating the accuracy of CDUS for GCA, using TAB as the gold standard, found a sensitivity and specificity of 68% and 81% respectively [50] indicating that the excellent results of the TABUL study were not ubiquitous. Axillary artery (AA) US may also play a role in the diagnosis of GCA and identification of extracranial disease. Using FDG-PET as a gold standard for extracranial GCA, Hop and colleagues found that 8/13 patients with PET avid axillary arteries had positive AA halo signs as well [51]. Comparing the use of AA CDUS with TA alone, they found that AA identified an additional 8 patients (out of 41) that were diagnosed by AA only and would have been missed if only TA US were used. AA CDUS increased sensitivity for GCA from 52 to 71%, supporting a role for including AA CDUS.
To attempt to circumvent issues of operator-dependent interpretation, a recent retrospective study applied a deep learning algorithm, using semantic segmentation technique to 137 GCA suspects across three clinical centers, resulting in a specificity of 95%, but a sensitivity of only 60% [52]. Greater data acquisition and manipulation of the positivity threshold could improve these results in the future, allowing for artificial intelligence in CDUS based diagnosis of GCA. A recent subanalysis of the TABUL study cohort demonstrated that the presence of the halo signs in multiple arterial segments of the TA increased the odds ratio of GCA diagnosis [53]. The same study demonstrated that the presence of the halo sign is associated with a positive TAB, and clinical symptoms such as jaw claudication and visual symptoms, but not with less specific ones such as headache and constitutional symptom. Interestingly, they also found that the presence of a positive halo on one side was associated with positive physical findings on exam, including thickening, tenderness and reduced pulse of that artery. More surprising however, was the finding that the rate of ipsilateral AION was increased (OR 4.7 right side, 6.4 left side) which refutes the dogma that ischemic visual symptoms do not correlate with the side of TA abnormalities [54]. Van der Geest and colleagues took this one step further, devising a composite halo score based on the degree of vessel wall thickness and found that both halo counts (numbers of involved segments) and the halo score were predictive of positive TABs, and were associated with risk of ischemic optic neuropathy. (OR 12.000 for a halo count ≥ 2 and OR 9.88 for a Halo Score ≥ 3) [55].
In 2018, the European League Against Rheumatism (EULAR) published recommendations for imaging in the diagnosis of GCA, with CDUS as the first line study owing to its specificity and fast response time as compared with TAB, which they recommended only if the results of the CDUS were not clear [56, 57]. In summary, CDUS has a growing role in the diagnosis of GCA, especially when used adjuvant in the setting of concurrent TAB.
MRI
Evidence is mounting for the utility of additional imaging techniques in the diagnosis of GCA such as MRI and FDG-PET. Using high field 3T post-contrast T1-weighted spin echo MRI of the scalp arteries with a grading system that takes into account mural wall thickening, enhancement and perivascular enhancement, Rhéaume and colleagues demonstrated a sensitivity of 93.6% and specificity of 77.9%, when using TAB as the gold standard [58]. The study also illustrated a negative predictive value for normal MRI of 94%, thus arguing that MRI may be used as a screening tool to see if TAB is necessary. A more recent study assessed the use of 3D fat saturated contrast enhanced vessel wall (CE VW) sequences vs. axial only 2D sequences [59]. Using a combination of +TAB and outside clinical judgment based on the ACR criteria as the gold standard, the authors found that CE VW had an 80% sensitivity and 100% specificity vs. the 70% sensitivity and 85% specificity of 2D vessel wall imaging. One of the advantages of this type of MRI is the ability to align the imaging along the plane of the course of the vessel so the wall can be imaged for its entire extent. Using 3D black blood (which suppresses mural blood signal so that mural wall enhancement can more easily be observed) T1 MRI, Rodriguez-Régent also demonstrated a sensitivity and specificity of 80% and 100% respectively [60]. By aligning the post-contrast imaging with time of flight (TOF) 2D MR angiogram images, arteries were better discernible from veins. The importance of this cannot be overstated, since mistaking extracranial veins for arteries can result in false positive errors. Importantly, this technique identified GCA in two cases where the TAB missed the diagnosis, and in two cases that were negative for GCA, the MRI allowed diagnosis of non-GCA central nervous system vasculitis.
Fluorine-18-fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT (FDG-PET) has gained traction as a diagnostic tool for GCA, since the inflammatory cells (macrophages and lymphocytes) that infiltrate vessel walls demonstrate high uptake of FDG. A recent comparison with TAB demonstrated a sensitivity and specificity of 92% and 85% respectively [61, 62•]. It also may help detect the diagnosis in cases where the primary symptoms are in the limbs, although TAB is still required to confirm GCA [63] and can detect aortitis in both cranial GCA and large vessel GCA [64]. While the primary role of FDG-PET in GCA diagnosis has been the identification of large vessel inflammation, a recent study has demonstrated utility in assessment of medium-sized intracranial arteries [65]. In this study, 24 patients with biopsy-proven GCA were compared with 24 controls using iterative reconstruction that optimized visualization of the temporal, occipital, maxillary and vertebral arteries. Using visual assessment by the radiologist, the sensitivity and specificity for GCA were 83% and 75% respectively; by analyzing the standardized uptake value (SUV) within a volume of interest, the sensitivity was similar at 79% but the specificity increased to 92% (using a cutoff of 5.00 for the SUVmax). The role of FDG-PET in GCA diagnosis is growing, but its high price tag, specific expertise required for analysis and radiation exposure renders it the least favorable for use in diagnostic imaging for GCA.
While TAB remains an important part of GCA work up, exact methodology still remains controversial. In a meta-analysis of 32 studies, from 12 countries, a sensitivity of 77% was found, when compared with diagnosis made by ACR criteria [66]. False negative results may result from either failure to find areas of location due to patchy involvement, or from lack of TA involvement in certain cases. Dermawan and colleagues evaluated 75 consecutive TABs from 2004 and found that of 62 that were found to be negative initially, four harbored pathological evidence of the disease when deeper levels of the original paraffin block were examined (median 228 levels vs. 8 originally examined) [67]. While none of the four patients (who either were not started on steroids or were tapered off) were known to lose vision, two did have recurrent symptoms when off steroids. These results indicate that a more comprehensive search for inflammation in pathological specimens would help reduce the false negative rate of TABs and potentially avoid cases of irreversible blindness.
In patients > 50 years old presenting with anterior ischemic optic neuropathy, where symptoms are not suggestive of GCA and serum markers are inconclusive, there is value in adjuvant ophthalmological testing to differentiate arteritic cases (AAION) from non-arteritic. Fluorescein angiography (FA) has been shown to reveal delayed retinal artery and/or choroidal filling in patients with arteritic AION (but not non-arteritic) and even in some cases of GCA without visual symptoms [68] but is invasive. Optical coherence tomography angiography (OCTa) is an emerging application of OCT that non-invasively images capillary perfusion at various levels of the retina. A recent 4 patient case series demonstrated dilation and eventual attenuation of superficial peripapillary capillaries in eyes with AAION, corresponding with visual field loss [69]. Larger series are needed to see if such a finding can be used to differentiate such cases from non-arteritic AION. The increased use of imaging modalities as an adjunctive or diagnostic complement to TAB has proven both valuable and efficient. Overall, utilizing the revised ACR criteria for GCA in conjunction with TAB and vascular imaging results in a high sensitivity for the disease. While we feel that TAB remains the gold standard since it reveals the actual pathology of the disease, we recommend using one vascular imaging technique as well to increase accuracy and to allow evaluation of other extracranial and intracranial arteries that might be affected by either GCA or alternative vasculitides.
Treatment: medications
Corticosteroids
The goal in GCA management is the reduction of ongoing inflammation and prevention of ischemic organ damage. Reduction of serum acute phase reactants such as ESR and CRP can be used a marker of successful control of inflammation but does not always correlate with relapses. Systemic corticosteroids (CS) remain the mainstay of initial treatment of GCA and it has been shown that vision loss is less likely to occur after they have been started [70]. While there is no consensus on the starting or maintenance dose of CS, it is universally accepted that when GCA is suspected, that high doses of systemic CS be started while awaiting TAB results. In cases where there is current or impending vision loss (e.g., transient blurred or double vision in setting of elevated ESR or CRP), pulsed IV methylprednisolone 1000 mg/day for 3 days is recommended, followed by a maintenance dose of 1 mg/kg of prednisone or equivalent [71]. However, in one study, visual outcomes were no worse when oral steroids were started promptly, as opposed to IV steroids [72]. However, speed of treatment has been showed to improve outcomes [73] and using IV steroids may allow faster achievement of therapeutic levels. The high relapse rate of GCA and the high morbidity associated with long-term CS use has prompted the need for steroid-sparing agents. Multiple immunosuppressants have been studied including biologic agents over the last few years.
Methotrexate
Methotrexate has long been used as an adjunctive immunodulator in GCA despite variable evidence for its use. A 4 year multicenter, randomized, double blind study compared treatment with 0.15 mg/kg/week MTX (increased to 0.25 mg/kg/week, for a maximum weekly dosage of 15 mg) or placebo, plus weight-based prednisone and found that there was no difference in the incidence of treatment failure between the two groups at 12 months, serious morbidity due to the disease, cumulative steroid doses or treatment toxicity [74]. However, in a meta-analysis of three randomized placebo-controlled trials treating newly diagnosed GCA patients, methotrexate < 15 mg/week reduced the risk of first and second relapse by 35% and 51% respectively, while also reducing the cumulative doses of CS [75]. A subsequent observational study of 186 patients reported a reduction in relapse rate of 72% in the methotrexate group vs. steroids only group [76].
Finally, a more recent retrospective study of patients treated between 1998 and 2013 demonstrated that the addition of methotrexate in addition to CS reduced the relapse rate by 3x as much as steroids alone [77]. However, this finding was likely confounded by the fact that the treatment group started off with a much higher relapse rate (11.8 relapses/10 person years) than the CS alone group (4.45 relapses/10 person years). Furthermore, the cumulative CS dose was no lower in the treatment group. Side effects included fatigue (n = 1), gastrointestinal intolerance (n = 1), dizziness (n = 1), myelosuppression (n = 1), and drug interaction (n = 1) but it was generally well tolerated. These results suggest that methotrexate may be useful as an adjuvant treatment for GCA patients with a high relapse rate during steroid tapering.
Tocilizumab
The pro-inflammatory cytokine IL-6 appears to play a pivotal role in the pathogenesis of GCA. Serum levels of IL-6 have been shown to be elevated in untreated GCA patients relative to controls [9], to correlate with disease activity and to lower in response to steroid treatment. In fact, in one study, 89% of disease recurrences were associated with an elevation in IL-6, as compared with 58% that were associated with elevated ESR [78]. As such, it emerged as a rational candidate for non-steroidal GCA therapy.
Tocilizumab is a monoclonal antibody against both the soluble and membrane bound forms of the IL-6 receptor, thus inhibiting IL-6 receptor-dependent signal transduction, that has been approved for the treatment of rheumatoid arthritis in 2010 [79]. It is typically given at a dose of 4–8 mg/kg infusion once a month. The first documented use of tocilizumab to treat GCA was in a 2011 case report in which use of the drug resulted in normalization of inflammatory markers and allowed reduction in steroid dose [80]. Multiple case series followed [81, 82] with satisfactory clinical and serological responses, culminating in a report of 7 cases of GCA treated with the tocilizumab in which all achieved and maintained remission and were able to lower prednisone from a mean of 20.8 mg/day to 4.1 mg/day [83]. Complications included mild neutropenia, transaminitis, and one post-operative myocardial infarction, in a patient who still had medium and large vessel vasculitis on autopsy, despite treatment. A 2014 retrospective multicenter open-label study in Spain, in which 22 GCA patients were treated with the drug, demonstrated a persistent remission in 19 (86%), although complications (severe neutropenia, recurrent pneumonia, and cytomegalovirus infection) led to cessation in three patients, and one died from a stroke related to infectious endocarditis after the second infusion [84]. In a single center, phase 2, randomized, double-blind, placebo-controlled trial in Switzerland, subjects were given monthly infusions over 13 months, in addition to a prednisolone taper beginning at 1 mg/kg and compared with those who received placebo plus steroids [85]. Of 20 patients given tocilizumab, 17(85%) reached complete remission by week 12 vs. 4/10 (40%) of ten patients given placebo plus steroids. There were more severe adverse events in the placebo/steroid group (10 events/10 patients) than the tocilizumab/steroids group (7/20), but it is notable that within the tocilizumab group, there were 10 cases of infectious disease including an ocular infection (vs. 1 in the placebo group), and one case of Stevens-Johnson syndrome 3 days after an infusion. In a French study of 34 patients treated with tocilizumab for GCA, 20 of which had previously been treated with other non-steroidal agents, 28 experienced a marked improvement in the disease, although notably, a patient who had a CRAO experienced no improvement in vision [86]. Complications included neutropenia in 3, tuberculous pericarditis in 1, and most significantly, fatal septic shock in another.
The most convincing evidence for the use of tocilizumab in the treatment of GCA comes from the GiACTA trial [87•], which led to FDA approval in May, 2017. In this randomized, double blind, placebo-controlled trial, the 251 patients were randomized in a 2:1:1:1 fashion to receive, over a year’s period, subcutaneous tocilizumab 162 mg weekly with a prednisone taper (n = 100), every other week with a prednisone taper (n = 50), placebo with a prednisone taper over 52 weeks (n = 51) or placebo with a prednisone taper over 26 weeks (n = 50). The primary outcome was sustained remission (SR) of GCA, which was defined as the absence of flare and the normalization of the CRP concentration to less than 1 mg per deciliter, both of which were sustained from week 12 to week 52 while adhering to the prednisone taper. The authors found that 56% of those receiving tocilizumab weekly and 53% of those receiving it every other week achieved SR, despite adhering to the prednisone taper, while only 14% of those in the placebo group with a 26-week taper and 18% of those in the placebo group with a 52-week taper achieved SR. Total median prednisone doses were reduced significantly in the treatment group: 1862 mg in both groups that received tocilizumab, as compared with 3296 mg in the 26-week prednisone taper placebo group and 3818 mg in the 52-week taper placebo group. In the two-year extension study of the GiACTA trial, approximately 47% of the patients who received Tocilizumab were able to maintain their remission over the additional two-year period [88]. Patients receiving tocilizumab had lower cumulative dose (2647 and 3782 in the weekly and biweekly Tocilizumab group vs 5248 and 5323 in the 26-week and 52-week taper placebo groups respectively) and experienced less adverse events. Quality of life assessment using the F-36 physical component tool showed that the group that received tocilizumab weekly scored 5.59 points higher than the 52-week taper placebo group. Serious adverse events were less common in the treatment groups (15% in the weekly group and 14% in the every other week group) vs. the placebo groups (22% in the 26-week taper placebo group and 25% in the 52-week taper placebo group). However, withdrawal from the trial due to an adverse event was slightly more common in the treatment groups (6%) vs. the placebo groups (4% in the 26-week taper group and 0% in the 52-week taper group). Furthermore, 4% of the tocilizumab patients experienced grade III neutropenia. It is noteworthy that only one patient in the trial experienced an AAION during a flare up, and this patient was in the every other week tocilizumab group. Limitations of the study include the fact that only 57% of the tocilizumab weekly and 68% of the every other week group were confirmed by TAB. Furthermore, the definition of “flare” upon which sustained remission was based on, was investigator dependent, so that it difficult to determine whether the flare ups in the treatment and placebo groups were equivalent. Neuro-ophthalmic data was not provided in the study, so that differences in asymptomatic optic nerve or retinal ischemia may have been missed. Finally, the 26-week taper group reflects an aggressive tapering regimen that is unlikely to result in an SR. A small Swiss study showed normalization of vessel wall enhancement on MRA in only a slightly greater percentage of patients (33%) receiving tocilizumab + steroids as compared with placebo + steroids (25%) although all tocilizumab patients experienced clinical and laboratory remission [89]. It should also be noted that the expense of tocilizumab far outweighs that of corticosteroids, costing from $25,000–40,000/year [90], and it requires a weekly subcutaneous injection, which may be difficult for some elderly patients and their families to administer at home. Despite these limitations, the results of the study support the use of tocilizumab over a year, in the long-term management of GCA, allowing for a higher chance of sustained remission and reduced cumulative steroid doses. The practitioner utilizing it must monitor for neutropenia, and must be aware that patients must still be monitored carefully for vision-threatening ischemic events that require emergent pulsed steroids along the way, since nearly half the treated patients still experienced relapses over the course of the study. Our practice is in line with the recent recommendations by the EULAR—to treat GCA urgently with corticosteroids, and to add adjunctive therapy with tocilizumab in patients with refractory or relapsing disease, or in whom a risk factor such as diabetes mellitus increases the risk of glucocorticoid-associated complications [91•].
Emerging therapies
Abatacept, a recombinant Ig-CTLA-4 fusion protein, binds to CD80/86 on activated dendritic or antigen presenting cells, inhibiting its interaction with CD28 and thereby preventing CD4+ T cell activation and IL-6 production. As a novel agent for GCA management, abatacept has been shown to maintain CS free remission for 9.9 months versus 3.9 months in patients receiving placebo. Relapse-free survival at 12 months with intravenous abatacept was 48% vs 31% in patients receiving placebo [92]. Furthermore, there were no differences in the frequency of severity of adverse effects between abatacept vs placebo treated groups. The study was limited by the relatively low number of subjects (n = 41). Another promising biological agent is ustekinumab, a monoclonal antibody that targets interleukin-12 and interleukin-23 and disrupts the immune response of Th1 and Th17, respectively. In a prospective, open-label study on 25 patients with refractory GCA on chronic CS, ustekinumab was associated with a reduction of the median cumulative CS dose from 5475 mg prior to ustekinumab use to 2790 mg after ustekinumab therapy at 1 year without relapse, but this study is limited by its small numbers [93]. A follow-up study demonstrated a reduction in median daily steroid dose from 20 mg of prednisolone to 5 mg by 52 weeks [94], but such a decrease would not be unexpected after a year even without the medication in our experience. While both abatacept and ustekinumab have shown intriguing results, additional clinical trials are needed to provide further insight into whether these agents could be used as either an add on or replacement of tocilizumab for management of GCA.
Leflunomide is a pyrimidine synthesis inhibitor that blocks T cell expansion and has been used in the treatment in rheumatoid arthritis and certain vasculitides. A recent prospective, observational patient choice study comparing steroids with leflunomide to steroids alone found that during the first 48 weeks of follow-up, out of 22 patients who relapsed, 4 (13.3%) were in the leflunomide group and 18 (39.1%) in the glucocorticoid-only group [95].
Antiviral treatment
The use of acyclovir or valacyclovir in patients with GCA is unsubstantiated, though speculation of VZV as a causative agent of GCA as discussed above has prompted the rare use of these medications in steroid refractory patients with the disease. Gilden et al. reported clinical improvement in an elderly female presenting with symptoms suggestive of GCA and including temporal pain and jaw claudication, along with symptoms of Takayasu’s arteritis such as loss of upper extremity pulses [96]. Treatment with intravenous acyclovir for 2 weeks, followed by oral valacyclovir led to dramatic improvement. However, as both the ESR and TAB were negative, and the CRP of 1.6 mg/dL was normal for age, we do not see any clear evidence that this was truly a case of GCA. As of this writing we were not able to find any studies on the effect of adjuvant treatment with valacyclovir or acyclovir on outcomes in GCA. Studies are needed to ascertain whether it confers any benefit at all in patients with GCA and we cannot recommend its use in typical cases at this time.
Aspirin
Ischemic stroke is a serious but uncommon complication of GCA, reported in up to 7% of patients [97]. The use of aspirin in the prevention of ischemic injury has been debated. A recent Israeli study looked at 136 patients with GCA and found that survival was shorter than expected for age in 63% of the patients, especially those younger than 70 years of age at the time of diagnosis [98•]. Moreover, they found that vision loss from GCA lowered by 4.6 years as compared with those without vision loss. However, they found that treatment with low dose aspirin 100 mg/day led to a reduction in early mortality (hazard ratio of 0.62) and an increase in the 2-year survival from 66 to 90%. This study was limited by its retrospective, non-controlled design. Nevertheless, it suggests that aspirin confers a benefit on patients with GCA. Therefore, if no contraindications exist, clinicians should consider adding low dose aspirin when managing patients with GCA. Practitioners must be aware that the addition of aspirin to steroid treatment can increase the risk of gastritis.
Treatment: surgery
There is no surgical treatment for GCA. The role of TAB in its diagnosis is discussed above.
Treatment: diet and lifestyle
There are no dietary or lifestyle changes that have been proven to improve the risk of complications from giant cell arteritis. However, patients on corticosteroids should be counseled to exercise and avoid high salt intake, to ameliorate the weight gain and fluid accumulation association with the medication. Weight bearing exercises, bone density monitoring and intake of vitamin D and calcium may help reduce the risk of osteoporosis and fractures.
See Table 1 for a summary of treatment options in GCA.
Conclusion
Despite major advances in our understanding of the immunologic and pathological underpinnings of GCA, including suggestions of an environmental or infectious trigger, its initial cause remains unclear. New diagnostic strategies, including temporal artery ultrasound, MRI, and PET scans, can complement or in some cases even replace TAB, but our ability to diagnose the disease with complete accuracy remains imperfect. Finally, the long-trusted use of corticosteroids in the treatment of GCA has been buttressed by the addition of steroid-sparing agents, most notably the IL-6 inhibitor tocilizumab, but even still, the disease remains a fierce adversary, capable of threatening both vision and life.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Dumont A, Lecannuet A, Boutemy J, Maigné G, Martin-Silva N, Deshayes S, et al. Characteristics and outcomes of patients with ophthalmologic involvement in giant-cell arteritis: a case-control study. Semin Arthritis Rheum. 2020;50(2):335–41. https://doi.org/10.1016/j.semarthrit.2019.09.008.
Horton BT, Magath TB, Brown GE. An undescribed form of arteritis of the temporal vessels. Proc Staff Meet Mayo Clinic. 1932;7:700–1.
Hutchinson J. Diseases of the arteries, I.–on a peculiar form of thrombotic arteritis of the aged which is sometimes productive of gangrene. Arch Surg (London) 1890;1:323–329.
Dequeker JV. Polymyalgia rheumatica with temporal arteritis, as painted by Jan van Eyck in 1436. Can Med Assoc J. 1981;124:1597–8.
Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199:173–83.
Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012;64:3788–98.
Weyand CM, Younge BR, Goronzy JJ. IFN-γ and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol. 2011;23(1):43–9. https://doi.org/10.1097/BOR.0b013e32833ee946.
Lozano E, Segarra M, García-Martínez A, Hernández-Rodríguez J, Cid MC. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis. 2008;67(11):1581–8. https://doi.org/10.1136/ard.2007.070805.
Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36:1286–94.
Al-Abdulla NA, Rismondo V, Minkowski JS, Miller NR. Herpes zoster vasculitis presenting as giant cell arteritis with bilateral internuclear ophthalmoplegia. Am J Ophthalmol. 2002;134:912–4.
Gilden D, White T, Khmeleva N, et al. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology. 2015;84(19):1948–1955 This controversial study suggested a role of VZV infection in the pathophysiology of GCA, based on an analysis of temporal artery vessel wall pathology using thinner slicing than is typically performed. Questions remain as to the specificity of the stain used and others have not been able to reproduce the findings as of yet.
Pisapia DJ, Lavi E. VZV, temporal arteritis, and clinical practice: false positive immunohistochemical detection due to antibody cross-reactivity. Exp Mol Pathol. 2016 Feb;100(1):114–5.
Sammel AM, Smith S, Nguyen K, et al. Assessment for varicella zoster virus in patients newly suspected of having giant cell arteritis [published online ahead of print, 2019 Nov 27]. Rheumatology (Oxford). 2019;kez556. doi:https://doi.org/10.1093/rheumatology/kez556
Solomon IH, Docken WP, Padera RF Jr. Investigating the association of giant cell arteritis with varicella zoster virus in temporal artery biopsies or ascending aortic resections. J Rheumatol. 2019;46(12):1614–8. https://doi.org/10.3899/jrheum.180912.
Giardina A, Rizzo A, Ferrante A, Capra G, Triolo G, Ciccia F. Giant cell arteritis associated with chronic active Epstein-Barr virus infection. Reumatismo. 2013 Mar 28;65(1):36–9.
Rodriguez-Pla A, Bosch-Gil JA, Echevarria-Mayo JE, Rossello-Urgell J, Solans-Laque R, Huguet-Redecilla P, et al. No detection of parvovirus B19 or herpesvirus DNA in giant cell arteritis. J Clin Virol. 2004 Sep;31(1):11–5.
Stamatis P, Turkiewicz A, Englund M, et al. Infections are associated with increased risk of giant cell arteritis-a population-based case-control study from Southern Sweden [published online ahead of print, 2020 May 15]. J Rheumatol. 2020;jrheum.200211. doi:https://doi.org/10.3899/jrheum.200211
Gonzalez-Gay MA, Vazquez-Rodriguez TR, Lopez-Diaz MJ, Miranda-Filloy JA, Gonzalez-Juanatey C, Martin J, et al. Epidemiology of giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2009;61:1454–61.
Bara L, Pope JE, Pequeno P, et al. Incidence and prevalence of giant cell arteritis in Ontario, Canada [published online ahead of print, 2020 Apr 6]. Rheumatology (Oxford). 2020;keaa095. doi:https://doi.org/10.1093/rheumatology/keaa095
Salvarani C, Crowson CS, O’Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. 2004;51(2):264–8. https://doi.org/10.1002/art.20227.
Haugeberg G, Paulsen PQ, Bie RB. Temporal arteritis in Vest Agder County in southern Norway: incidence and clinical findings. J Rheumatol. 2000;27(11):2624–7.
Gonzalez-Gay MA, Miranda-Filloy JA, Lopez-Diaz MJ, Perez-Alvarez R, Gonzalez-Juanatey C, Sanchez-Andrade A, et al. Giant cell arteritis in northwestern Spain: a 25-year epidemiologic study. Medicine (Baltimore). 2007;86(2):61–8. https://doi.org/10.1097/md.0b013e31803d1764.
Muratore F, Crowson CS, Boiardi L, et al. Comparison of biopsy-proven giant cell arteritis in North America and Southern Europe: a population-based study. Clin Exp Rheumatol. 2020;38 Suppl 124(2):79–83.
Bas-Lando M, Breuer GS, Berkun Y, Mates M, Sonnenblick M, Nesher G. The incidence of giant cell arteritis in Jerusalem over a 25-year period: annual and seasonal fluctuations. Clin Exp Rheumatol. 2007;25(1 Suppl 44):S15–7.
Yates M, Luben R, Hayat S, et al. Cardiovascular risk factors associated with polymyalgia rheumatica and giant cell arteritis in a prospective cohort: EPIC-Norfolk Study. Rheumatology (Oxford). 2020;59(2):319–323. doi:https://doi.org/10.1093/rheumatology/kez289. This study demonstrates that hypertension increases the risk of GCA while the following study suggests a negative association with blood glucose levels, together advancing our understanding of vascular risk factors for the disease.
Wadström K, Jacobsson L, Mohammad AJ, Warrington KJ, Matteson EL, Turesson C. Negative associations for fasting blood glucose, cholesterol and triglyceride levels with the development of giant cell arteritis [published online ahead of print, 2020 Apr 2]. Rheumatology (Oxford). 2020;keaa080. doi:https://doi.org/10.1093/rheumatology/keaa080
Andersen JB, Myklebust G, Haugeberg G, Pripp AH, Diamantopoulos AP. Incidence trends and mortality of giant cell arteritis in Southern Norway [published online ahead of print, 2020 Jan 7]. Arthritis Care Res (Hoboken). 2020;https://doi.org/10.1002/acr.24133. doi:https://doi.org/10.1002/acr.24133
Aouba A, Gonzalez Chiappe S, Eb M, Delmas C, de Boysson H, Bienvenu B, et al. Mortality causes and trends associated with giant cell arteritis: analysis of the French national death certificate database (1980-2011). Rheumatology (Oxford). 2018;57(6):1047–55. https://doi.org/10.1093/rheumatology/key028.
Font C, Cid MC, Coll-Vinent B, López-Soto A, Grau JM. Clinical features in patients with permanent visual loss due to biopsy-proven giant cell arteritis. Br J Rheumatol. 1997;36(2):251–4. https://doi.org/10.1093/rheumatology/36.2.251.
Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125(4):509–20. https://doi.org/10.1016/s0002-9394(99)80192-5.
Mednick Z, Farmer J, Khan Z, Warder D, Ten Hove M. Coronary arteritis: an entity to be considered in giant cell arteritis. Can J Ophthalmol. 2016;51(1):e6–8. https://doi.org/10.1016/j.jcjo.2015.09.011.
Morvai-Illés B, Ágoston G, Séllei Á, Kovács L, Varga A. Giant cell arteritis presenting with pericardial effusion, hoarseness, and amaurosis. Anatol J Cardiol. 2020;23(4):235–237. doi:https://doi.org/10.14744/AnatolJCardiol.2019.00502
Truffaut L, Lefebvre P. Tongue necrosis in Giant-cell arteritis. N Engl J Med. 2018;378(26):2517. https://doi.org/10.1056/NEJMicm1709412.
Konishi C, Nakagawa K, Nakai E, Nishi K, Ishikawa R, Uematsu S, et al. Interstitial lung disease as an initial manifestation of giant cell arteritis. Intern Med. 2017;56(19):2633–7. https://doi.org/10.2169/internalmedicine.8861-17.
Clark AE, Victor WH. An unusual presentation of temporal arteritis. Ann Ophthalmol. 1987 Sep;19(9):343–6.
Mitchell JR, Krashin-Bichler I, Rosenblum M, Diamond EL, Dinkin MJ. Giant cell arteritis presenting with bilateral orbital inflammatory disease and enhancing superficial temporal arteries. Pract Neurol. 2014;14(6):446–7.
Rezaei S, Prospero Ponce CM, Vickers A, Divatia M, Lee AG. Giant cell arteritis relapse presenting as idiopathic orbital inflammation. Can J Ophthalmol. 2020 Feb;55(1):e36–9.
Hayreh SS, Podhajsky PA, Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998;125(4):521–6.
Levin F, Schubert HD, Merriam JC, Blume RS, Odel JG. Occult temporal arteritis in a 54-year-old man. J Neuroophthalmol. 2011 Jun;31(2):153–4.
Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122–8.
Chan FLY, Lester S, Whittle SL, Hill CL. The utility of ESR, CRP and platelets in the diagnosis of GCA. BMC Rheumatol. 2019;3:14. Published 2019 Apr 10.
Salehi-Abari I. 2016 ACR revised criteria for early diagnosis of giant cell (temporal) arteritis. Autoimmune Dis Ther Approaches Open Access. 2016;3:1–4.
Niederkohr RD, Levin LA. A Bayesian analysis of the true sensitivity of a temporal artery biopsy. Invest Ophthalmol Vis Sci. 2007;48(2):675–80. https://doi.org/10.1167/iovs.06-1106.
Achkar AA, Lie JT, Hunder GG, O’Fallon WM, Gabriel SE. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med. 1994;120(12):987–92. https://doi.org/10.7326/0003-4819-120-12-199406150-00003.
Chung SH, Morcos MB, Ng B. Determinants of positive temporal artery biopsies in the Veterans Health Administration National Database Cohort. Arthritis Care Res (Hoboken). 2020;72(5):699–704. doi:https://doi.org/10.1002/acr.23897. Based on this large review of temporal artery biopsies, we should be obtaining bilateral specimens of 3.0 cm or greater to improve our sensitivity, regardless of duration of prior prednisone therapy.
Chakrabarty A, Mackie S, Harden C, Morgan AW. Temporal artery biopsy: audit of histological diagnosis. Rheumatology (Oxford). 2020;59(3):678–9. https://doi.org/10.1093/rheumatology/kez396.
Chrysidis S, Duftner C, Dejaco C, Schäfer VS, Ramiro S, Carrara G, et al. Definitions and reliability assessment of elementary ultrasound lesions in giant cell arteritis: a study from the OMERACT large vessel vasculitis ultrasound working group. RMD Open. 2018;4:e000598–9.
Luqmani R, Lee E, Singh S et al. The role of ultrasound compared to biopsy of temporal arteries in the diagnosis and treatment of Giant Cell Arteritis (TABUL): a diagnostic accuracy and cost-effectiveness study. Health Technol Assess 2016;20:1–238. The TABUL (Temporal Artery Biopsy vs ULtrasound in Diagnosis of GCA) demonstrated that the halo sign, a result of vessel wall edema and intimal thickness, had a similar sensitivity compared to temporal artery biopsy (TAB) (93% vs. 91%), although it was less specific (77% vs. 81%).
Fernández-Fernández E, Monjo-Henry I, Bonilla G, Plasencia C, Miranda-CarúsME, Balsa A, De Miguel E. False positives in the ultrasound diagnosis of giant cell arteritis: some diseases can also show the halo sign. Rheumatology(Oxford). 2020.
Rinagel M, Chatelus E, Jousse-Joulin S, Sibilia J, Gottenberg JE, Chasset F, et al. Diagnostic performance of temporal artery ultrasound for the diagnosis of giant cell arteritis: a systematic review and meta-analysis of the literature. Autoimmun Rev. 2019;18(1):56–61.
Hop H, Mulder DJ, Sandovici M, Glaudemans AWJM, van Roon AM, Slart RHJA, Brouwer E. Diagnostic value of axillary artery ultrasound in patients with suspected giant cell arteritis. Rheumatology (Oxford). 2020 Apr 2:
Roncato C, Perez L, Brochet-Guégan A, Allix-Béguec C, Raimbeau A, Gautier G, Agard C, Ploton G, Moisselin S, Lorcerie F, Denis G, Gombert B, Gervais E, Espitia O. Colour Doppler ultrasound of temporal arteries for the diagnosis of giant cell arteritis: a multicentre deep learning study. Clin Exp Rheumatol. 2020 Mar-Apr;38 Suppl 124(2):120–125.
Ponte C, Serafim AS, Monti S, Fernandes E, Lee E, Singh S, Piper J, Hutchings A, McNally E, Diamantopoulos AP, Dasgupta B, Schmidt WA, Luqmani RA. Early variation of ultrasound halo sign with treatment and relation with clinical features in patients with giant cell arteritis. Rheumatology (Oxford). 2020 May 11:keaa196.
Schmidt WA, Krause A, Schicke B, Kuchenbecker J, Gromnica-Ihle E. Do temporal artery duplex ultrasound findings correlate with ophthalmic complications in giant cell arteritis? Rheumatology. 2009;48:383–5.
van der Geest KSM, Borg F, Kayani A, Paap D, Gondo P, Schmidt W, et al. Novel ultrasonographic Halo Score for giant cell arteritis: assessment of diagnostic accuracy and association with ocular ischaemia. Ann Rheum Dis. 2020 Mar;79(3):393–9.
Dejaco C, Ramiro S, Duftner C, Besson FL, Bley TA, Blockmans D, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis. 2018;77(5):636–43.
Monti S, Águeda AF, Luqmani RA, Buttgereit F, Cid M, Dejaco C, et al. Systematic literature review informing the 2018 update of the EULAR recommendation for the management of large vessel vasculitis: focus on giant cell arteritis. RMD Open. 2019;5(2):e001003.
Rhéaume M, Rebello R, Pagnoux C, Carette S, Clements-Baker M, Cohen-Hallaleh V, et al. High-resolution magnetic resonance imaging of scalp arteries for the diagnosis of giant cell arteritis: results of a prospective cohort study. Arthritis Rheumatol. 2017;69(1):161–8. https://doi.org/10.1002/art.39824.
Poillon G, Collin A, Benhamou Y, Clavel G, Savatovsky J, Pinson C, et al. Increased diagnostic accuracy of giant cell arteritis using three-dimensional fat-saturated contrast-enhanced vessel-wall magnetic resonance imaging at 3 T. Eur Radiol. 2020 Apr;30(4):1866–75.
Rodriguez-Régent C, Ben Hassen W, Seners P, Oppenheim C, Régent A. 3D T1-weighted black-blood magnetic resonance imaging for the diagnosis of giant cell arteritis. Clin Exp Rheumatol. 2020 Mar-Apr;38 Suppl 124(2):95–98.
Braun J, Baraliakos X, Fruth M. The role of 18F-FDG positron emission tomography for the diagnosis of vasculitides. Clin Exp Rheumatol. 2018;36 Suppl 114(5):108–114.
Sammel AM, Hsiao E, Schembri G, et al. Diagnostic Accuracy of positron emission tomography/computed tomography of the head, neck, and chest for giant cell arteritis: a prospective, double-blind, cross-sectional study. Arthritis Rheumatol. 2019;71(8):1319–1328. We do not yet use PET scans for the routine diagnosis of giant cell arteritis, but this study demonstrates a strong sensitivity for the disease that support its use in cases that are otherwise difficulty to diagnose.
Ahmadi Bidakhvidi N, Goffin K, Van Laere K, Gheysens O, Blockmans D. 18F-FDG PET in giant cell arteritis. Clin Nucl Med. 2020;45(2):170–1.
Bellan M, Puta E, Croce A, Sacchetti GM, Orsini F, Zecca E, et al. Role of positron emission tomography in the assessment of disease burden and risk of relapse in patients affected by giant cell arteritis. Clin Rheumatol. 2020;39(4):1277–81. https://doi.org/10.1007/s10067-019-04808-7.
Nienhuis PH, Sandovici M, Glaudemans AW, Slart RH, Brouwer E. Visual and semiquantitative assessment of cranial artery inflammation with FDG-PET/CT in giant cell arteritis [published online ahead of print, 2020 Jun 1]. Semin Arthritis Rheum. 2020;50(4):616–23.
Rubenstein E, Maldini C, Gonzalez-Chiappe S, Chevret S, Mahr A. Sensitivity of temporal artery biopsy in the diagnosis of giant cell arteritis: a systematic literature review and meta-analysis. Rheumatology (Oxford). 2020;59(5):1011–20.
Dermawan JK, Prayson RA. Evaluation of deeper levels in initially negative temporal artery biopsies and likelihood of a positive result. Ann Diagn Pathol. 2020 Apr 8;46:151517.
Siatkowski RM, Gass JD, Glaser JS, Smith JL, Schatz NJ, Schiffman J. Fluorescein angiography in the diagnosis of giant cell arteritis. Am J Ophthalmol. 1993;115(1):57–63. https://doi.org/10.1016/s0002-9394(14)73525-1.
Gaier ED, Gilbert AL, Cestari DM, Miller JB. Optical coherence tomographic angiography identifies peripapillary microvascular dilation and focal non-perfusion in giant cell arteritis. Br J Ophthalmol. 2018;102(8):1141–6. https://doi.org/10.1136/bjophthalmol-2017-310718.
Aiello PD, Trautmann JC, McPhee TJ, et al. Visual prognosis in giant cell arteritis. Ophthalmology. 1993;100:550–5.
Mackie SL, Dejaco C, Appenzeller S, et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis. Rheumatology (Oxford). 2020;59(3):e123. https://doi.org/10.1093/rheumatology/kez672.
Hayreh SS, Zimmerman B. Visual deterioration in giant cell arteritis patients while on high doses of corticosteroid therapy. Ophthalmology. 2003;110:1204–15.
Gonzales-Gay MA, Blanco R, Rodriguez-Valverde V, Martinez-Taboada M, Delgado-Rodriguez M, Figueroa M, et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum. 1998;41:1497–504.
Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46(5):1309–18. https://doi.org/10.1002/art.10262.
Mahr AD, Jover JA, Spiera RF, Hernández-García C, Fernández-Gutiérrez B, LaValley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789–97.
Leon L, Rodriguez-Rodriguez L, Morado I, et al. Treatment with methotrexate and risk of relapses in patients with giant cell arteritis in clinical practice. Clin Exp Rheumatol. 2018;36 Suppl 111(2):121–128.
Koster MJ, Yeruva K, Crowson CS, Muratore F, Labarca C, Warrington KJ. Efficacy of methotrexate in real-world management of giant cell arteritis: a case-control study. J Rheumatol. 2019;46:501–8.
Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43:1041–8.
Alten R, Maleitzke T. Tocilizumab: a novel humanized anti-interleukin 6 (IL-6) receptor antibody for the treatment of patients with non-RA systemic, inflammatory rheumatic diseases. Ann Med. 2013;45:357–63.
Christidis D, Jain S, Das GB. Successful use of tocilizumab in polymyalgic onset biopsy positive GCA with large vessel involvement. BMJ Case Rep. 2011 Jun;30:2011.
Sciascia S, Rossi D, Roccatello D. Interleukin 6 blockade as steroid-sparing treatment for 2 patients with giant cell arteritis. J Rheumatol. 2011;38:2080–1.
Salvarani C, Magnani L, Catanoso M, Pipitone N, Versari A, Dardani L, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford). 2012;51(1):151–6. https://doi.org/10.1093/rheumatology/ker296.
Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken). 2012;64(11):1720–9. https://doi.org/10.1002/acr.21750.
Loricera J, Blanco R, Hernández JL, Castañeda S, Mera A, Pérez-Pampín E, et al. Tocilizumab in giant cell arteritis: multicenter open-label study of 22 patients. Semin Arthritis Rheum. 2015;44(6):717–23. https://doi.org/10.1016/j.semarthrit.2014.12.005.
Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10031):1921–7. https://doi.org/10.1016/S0140-6736(16)00560-2.
Régent A, Redeker S, Deroux A, Kieffer P, Ly KH, Dougados M, et al. Tocilizumab in giant cell arteritis: a multicenter retrospective study of 34 patients. J Rheumatol. 2016;43(8):1547–52. https://doi.org/10.3899/jrheum.151252.
Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377(4):317–328. doi:https://doi.org/10.1056/NEJMoa1613849. The GiACTA trial was the first study to provide evidence for a steroid-sparing agent in the management of giant arteritis based on prospective, placebo-controlled design. It has revolutionized the way we treat giant cell arteritis, as the use of the IL-6 inhibitor tocilizumab could lead to a significant reduction of steroid usage (and side effects) in the disease.
Stone JH, Min Bao, Jian Han, Martin Aringer, Daniel Blockmans, Elisabeth Brouwer, Maria C. Cid, Bhaskar Dasgupta, Jürgen Rech, Carlo Salvarani, Robert Spiera, Sebastian Unizony. OP0140 long-term outcome of tocilizumab for patients with Giant cell arteritis: results from part 2 of the GiACTA trial. Ann Rheum Dis 2019;78:145–146.
Reichenbach S, Adler S, Bonel H, Cullmann JL, Kuchen S, Bütikofer L, et al. Magnetic resonance angiography in giant cell arteritis: results of a randomized controlled trial of tocilizumab in giant cell arteritis. Rheumatology (Oxford). 2018;57:982–6.
Sadun A, Gordon L. Should tocilizumab be used routinely in new patients with a diagnosis of giant cell arteritis? J Neuroophthalmol. 2020;40(1):117–21. https://doi.org/10.1097/WNO.0000000000000869.
Hellmich B, Agueda A, Monti S, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2020;79(1):19–30. doi:https://doi.org/10.1136/annrheumdis-2019-215672. The recommendations of the EULAR that tocilizumab should be considered an add-on drug in patients with refractory or relapsing disease, or in whom a risk factor such as diabetes mellitus increases the risk of glucocorticoid-associated complications have put the findings of the GiACTA trial into practice.
Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol. 2017;69(4):837–45. https://doi.org/10.1002/art.40044.
Conway R, O’Neill L, O’Flynn E, et al. Ustekinumab for the treatment of refractory giant cell arteritis. Ann Rheum Dis. 2016;75:1578–9.
Conway R, O’Neill L, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, et al. Ustekinumab for refractory giant cell arteritis: a prospective 52-week trial. Semin Arthritis Rheum. 2018;48(3):523–8. https://doi.org/10.1016/j.semarthrit.2018.04.004.
Hočevar A, Ješe R, Rotar Ž, Tomšič M. Does leflunomide have a role in giant cell arteritis? An open-label study. Clin Rheumatol. 2019;38(2):291–6. https://doi.org/10.1007/s10067-018-4232-x.
Gilden D, White TM, Nagae L, Gurdin WH, Boyer PJ, Nagel MA. Successful antiviral treatment of giant cell arteritis and Takayasu arteritis. JAMA Neurol. 2015 Aug;72(8):943–6. doi: https://doi.org/10.1001/jamaneurol.2015.0840. PMID: 26258736; PMCID: PMC5489065.
Samson M, Jacquin A, Audia S, Daubail B, Devilliers H, Petrella T, et al.. Stroke associated with giant cell arteritis: a population-based study.J Neurol Neurosurg Psychiatry. 2015; 86:216–221. doi: https://doi.org/10.1136/jnnp-2014-307614
Nesher G, Poltorak V, Hindi I, et al. Survival of patient with giant cell arteritis: Impact of vision loss and treatment with aspirin. Autoimmun Rev. 2019;18(8):831–834. doi:https://doi.org/10.1016/j.autrev.2019.06.003. This recent study suggests that aspirin may confer a benefit in patients with giant cell arteritis. This conclusion is logical given the known ischemic nature of the disease and theoretical role of thrombosis. Larger studies are needed to confirm their findings.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Cerebrovascular Disorders
Rights and permissions
About this article

Cite this article
Dinkin, M., Johnson, E. One Giant Step for Giant Cell Arteritis: Updates in Diagnosis and Treatment. Curr Treat Options Neurol 23, 6 (2021). https://doi.org/10.1007/s11940-020-00660-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s11940-020-00660-2