
Abstract
Purpose of Review
In the preceding decade, the management of metastatic cutaneous melanoma has been revolutionised with the development of highly effective therapies including immune checkpoint inhibitors (specifically CTLA-4 and PD-1 inhibitors) and targeted therapies (BRAF and MEK inhibitors). The role of chemotherapy in the contemporary management of melanoma is undefined.
Recent Findings
Extended analyses highlight substantially improved 5-year survival rates of approximately 50% in patients with metastatic melanoma treated with first-line therapies. However, most patients will progress on these first-line treatments. Sequencing of chemotherapy following failure of targeted and immunotherapies is associated with low objective response rates and short progression-free survival, and thus, meaningful benefits to patients are minimal.
Summary
Chemotherapy has limited utility in the contemporary management of cutaneous melanoma (with a few exceptions, discussed herein) and should not be the standard treatment sequence following failure of first-line therapies. Instead, enrolment onto clinical trials should be standard-of-care in these patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The treatment of metastatic cutaneous melanoma has been revolutionised in the last decade with the introduction of immune-activating and targeted therapies. Prior to their development, chemotherapy was the mainstay of melanoma treatment — with 5-year survival rates of approximately 6–10% in patients with stage IV disease [1].
Treatment with immunotherapy, specifically immune checkpoint inhibition (ICI), has significantly improved melanoma survival compared to the chemotherapy era — with 5-year survival rates for single-agent PD-1 inhibitors of 34–44% in stage IV disease [2, 3] and 52% when combined with the CTLA-4 inhibitor ipilimumab [3]. As such, the PD-1 inhibitors nivolumab or pembrolizumab, alone or in combination with ipilimumab, are recommended as first-line systemic treatment for metastatic melanoma by the American Society of Clinical Oncology [4], the National Comprehensive Cancer Network [5] and the European Society of Medical Oncology [6].
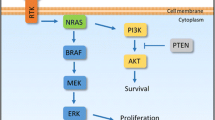
Approximately, 70% of melanoma have activating mutations in the MAPK pathway, two thirds of which in the BRAF gene and are thus targetable by BRAF inhibitors (BRAFi) including vemurafenib, dabrafenib and encorafenib. When used in combination with MEK inhibitors (MEKi) (cobimetinib, trametinib or binimetinib), which target downstream kinases in the MAPK pathway, 5-year survival for BRAF inhibition ranges from 31 to 35% [7,8,9]. Thus, guidelines [4,5,6] also propose MAPK-targeted therapy (TT) as an option for BRAF-mutant patients.
No guidelines explore the role of cytotoxic chemotherapy in the contemporary management of melanoma. Therefore, it is relevant to examine whether this prototypical anticancer therapy still has a role in the management of patients with metastatic cutaneous melanoma and in what specific contexts.
Chemotherapy in Melanoma
Table 1 summarises historical chemotherapy agents used for metastatic cutaneous melanoma, including clinical efficacy outcomes and toxicities.
Alkylating Agents
Dacarbazine (DTIC) was the first cytotoxic chemotherapeutic agent approved by the FDA (in 1975) for metastatic melanoma and prior to contemporary treatments was considered standard-of-care [38]. Dacarbazine is metabolised by the liver into the active metabolite MTIC, which forms DNA lesions such as O6-methylguanine resulting in double-stranded DNA breaks [39]. The overall response rate (ORR) to dacarbazine varies from 9 to 29% in the first-line (1L) setting; however, durable complete responses are rare (< 2%) [10]. A meta-analysis of 20 trials of dacarbazine identified a median OS of 7.4 months when used as 1L therapy [11]. Notably, no trials compare dacarbazine with best supportive care (BSC); and thus, meaningful benefits to patients are unclear — despite it having become the main comparator for trials in metastatic melanoma after having been proposed as such following a Cochrane review in 2000 [10].
Temozolomide is another alkylating agent given orally which is metabolised to MTIC. Contrary to dacarbazine, no prior hepatic conversion is required [40]. A systematic review of 26 studies of temozolomide in the 1L and beyond first-line (1L +) setting identified variable ORR from 9 to 29%, while median OS ranged from 4.0 to 13.1 months [12]. A phase III trial comparing temozolomide with dacarbazine demonstrated similar median OS (7.7 vs 6.4 months, respectively) and ORR (13.5% vs 12.1%, respectively) [13] as 1L therapy. Unlike dacarbazine, temozolomide is able to cross the blood–brain barrier and thus may have superior efficacy against central nervous system (CNS) metastases. One retrospective study of 122 patients with intracranial disease reported a 7% ORR (1L) [41], while the risk of CNS relapse was reduced by 77% with temozolomide versus dacarbazine in another retrospective study [42].
Fotemustine is a nitrosourea which, in addition to DNA and RNA alkylation leading to unstable target lesions such as O6-chlorethylguanine, also inhibits DNA repair enzymes such as DNA polymerase and nucleotidyltransferase [43]. A phase III study demonstrated superior ORR for fotemustine compared to dacarbazine (15.2% vs 6.8%, respectively, P = 0.043) in the 1L, although median OS was not significantly improved (7.3 months vs 6.4 months, respectively; P = 0.059) [14]. Due to its lipophilicity, fotemustine can penetrate the blood–brain barrier, with a retrospective cohort study of 36 patients with intracranial disease demonstrating an ORR of 25.0% [15].
Taxanes
Taxanes inhibit mitosis by enhancing tubulin polymerisation to stabilise microtubules and halting the cell cycle. Three phase II studies of paclitaxel (at 250 mg/m2/24 h every 21 days) in metastatic melanoma demonstrated ORRs of 12–36%, while median OS was not reported in any [16,17,18]. However, due to the high frequency of neutropenia, shorter-/lower-dose infusions (at 80 mg/m2/1 h every 7 days) are more commonly given [44]. Three phase II studies (all 1L +) reported ORRs of 0–15.6% and median OS of 7–7.8 months in melanoma with this regimen [19,20,21].
However, pre-medication with steroids (e.g. dexamethasone) and H2-antihistamines (e.g. famotidine) is frequently required to reduce hypersensitivity reactions (manifesting as hypotension, bronchospasm and angioedema) which occur in approximately 11% of patients [45]. These reactions are theorised to be secondary to paclitaxel itself or the polyethoxylated castor oil solvent (Cremophor® EL). Consequently, nanoparticle albumin-bound paclitaxel (Nab-paclitaxel) was developed without Cremophor and does not require pre-medication against hypersensitivity reactions while additionally demonstrating superior antitumour activity in vitro [46]. Nab-paclitaxel has been studied in one randomised phase III trial against dacarbazine in metastatic melanoma (1L) — with comparable ORRs (15 vs 11%, respectively, P = 0.24) and median OS (12.6 vs 10.5 months, P = 0.27) [22].
Another derivative taxane is docosahexanoic acid–paclitaxel (DHA-paclitaxel), consisting of a fatty acid (DHA)-paclitaxel conjugate. Murine pharmacokinetic studies highlight sixfold increased tumoural concentration by DHA-paclitaxel compared with Cremophor-bound paclitaxel [47]. DHA-paclitaxel has been compared in one phase III trial against dacarbazine in melanoma patients in the 1L setting — with comparable ORRs (5.2 vs 5.5%, respectively) and median OS (8.9 vs 7.5 months) [23].
Docetaxel is another taxane with approximately twice the potency of microtubule stabilisation compared to paclitaxel in vitro [48]. Single-agent docetaxel has been studied in three phase II trials in metastatic melanoma (both 1L and 1L +) — yielding ORRs of 5.7–7%, including two durable complete responses [24,25,26]. Median OS was only reported in two studies, ranging from 6.5 to 13 months.
Platinum Chemotherapy
Single-agent platinum-based chemotherapy (‘platin’) regimens have demonstrated some activity against metastatic melanoma. Platins impair DNA repair mechanisms via platinum ion cross-linkage with purine bases on the DNA, inducing DNA damage and apoptosis. Historical trials of cisplatin reported ORRs of 6–15% in metastatic melanoma in the 1L + setting; however, OS was not reported [27]. One phase II trial of cisplatin in 89 patients highlighted an ORR of 19.8% and median OS of 7.6 months, with efficacy and tolerability not significantly improved by the addition of amifostine (a myelo- and nephro-protective thiol derivative) [28]. Nephrotoxicity was unacceptably frequent in the cohort (19.2%) — with three life-threatening cases (3.4%) and one death as a result (1.1%).
Carboplatin is a newer generation platin characterised by increased half-life but decreased potency compared to cisplatin [49]. One phase II trial of 26 patients (1L +) observed partial responses in five patients (ORR 19.2%) with a median duration of response of 5 months; however, median OS was not provided [29]. Notably, only one case of renal impairment was reported (3.8%). This is consistent with the different safety profiles of carboplatin compared with cisplatin — with increased myelosuppression but decreased nephrotoxicity [49].
A phase II trial of cisplatin–carboplatin combination chemotherapy was conducted aiming to increase total platinum dosage while mitigating the different side effect profiles of the two agents. Fifteen patients who had progressed on dacarbazine were treated, yielding an ORR of 26.4% including two complete responses (13.3%), with a median OS of 12.5 months [30]. Unlike with high-dose single-agent cisplatin, renal impairment was uncommon (13%), with no cases severe or life-threatening.
Combination Chemotherapy Regimens
Combining chemotherapy agents targeting different aspects of the cancer cell cycle is standard for many cancers to overcome tumour resistance to cytotoxicity and optimise varying adverse event (AE) profiles of individual drugs. As such, numerous combination chemotherapy combinations have been trialled in metastatic melanoma [33, 34, 35, 37, 50] with varying degrees of efficacy (key combinations featured in Table1). However, there is no evidence that such combinations confer improved survival compared to single-agent dacarbazine — with a Cochrane review and meta-analysis in 2018 determining an OS hazard ratio (HR) of 0.99 (95%CI 0.85–1.16) for polychemotherapy versus single-agent chemotherapy, at the cost of increased treatment toxicities (HR 1.97, 95%CI 1.44–2.71) [51]. Many of such combinations utilise a dacarbazine backbone; however, a further meta-analysis of 20 randomised trials found no significantly improved survival between dacarbazine alone and dacarbazine-based polychemotherapy (HR 1.16, 95%CR 0.87–1.54) [11].
Notably, the sole prospective study comparing chemotherapy with BSC in melanoma evaluates a non-randomised multicentre cohort treated with either cisplatin, vinblastine and dacarbazine (CVD) or BSC as 1L treatment [35]. Initially planned for randomisation this was altered to ‘patient preference’ after inadequate recruitment for the BSC arm. While ORR and median OS were superior in patients receiving CVD compared to BSC (9.6 vs 0% and 137 vs 229 days respectively) this was confounded by greater frequency of CNS disease and worse performance status of the BSC arm at baseline and thus definitive benefit of chemotherapy could not be concluded. Furthermore there were no differences in quality-of-life (QoL) scoring between treatment arms after 8 weeks despite improved ORR and OS.
Chemotherapy Versus Contemporary Treatment Options
The approval of ipilimumab, the first ICI, soon followed by vemurafenib, the first TT, in 2011 heralded a paradigm shift in the treatment of melanoma [52]. These agents rapidly became standard-of-care replacing chemotherapy, with a trend towards improved survival [53•]. Relevant prospective randomised trials comparing such therapies versus chemotherapy are summarised in Table 2.
Chemotherapy Versus Immune Checkpoint Inhibition
One phase III randomised trial compared ipilimumab plus dacarbazine versus dacarbazine alone (1L), demonstrating similar ORR (15.2 vs 10.3%; P = 0.09) but improved median progression-free survival (PFS) (HR 0.76, P = 0.006) and OS (11.0 vs 8.9 months; HR 0.69, P < 0.001) [54]. This corresponded to superior 5-year survival rates in the ipilimumab plus dacarbazine group compared to dacarbazine alone (18.2% vs 8.8%, P = 0.002) [55]. However, 5-year survival of ipilimumab alone in other studies of untreated metastatic melanoma is approximately 26% [3], and thus, the value of added dacarbazine is unclear. AEs were slightly increased with the addition of ipilimumab compared to single-agent dacarbazine — with 98.8% vs 94.0% developing any grade events and 56.3% vs 27.5% for grade 3 or 4 events.
Tremelimumab, another CTLA-4 inhibitor, was compared against investigators’ choice chemotherapy (dacarbazine or temozolomide) in another randomised trial (1L) [56]. This showed no significant difference in ORR between the tremelimumab and chemotherapy cohorts (10.7 vs 9.8%, respectively; P = 0.09). While overall PFS was similar in both groups (6 months PFS rate 20.3 vs 18.1%; P = 0.48), for the patients who did respond to tremelimumab, their duration of response was significantly longer compared to chemotherapy responders (median 35.8 vs 13.7 months, P = 0.001). While this suggests CTLA-4 blockade provides sustained anticancer activity in a small subset of patients, no biomarkers to predict response were identified. Importantly, median OS was similar in both groups — 12.6 months in the tremelimumab arm and 10.7 in the chemotherapy arm (P = 0.13), suggesting chemotherapy is non-inferior to CTLA-4 blockade in unselected cohorts.
KEYNOTE-002 is a randomised trial comparing pembrolizumab with investigators’ choice chemotherapy (carboplatin–paclitaxel, paclitaxel, carboplatin, dacarbazine or temozolomide) in ipilimumab and BRAF/MEKi-refractory melanoma (1L +) [57]. Two doses of pembrolizumab (2 mg/kg and 10 mg/kg) were studied, both demonstrating ORRs superior to chemotherapy (21% and 25% vs 4%, respectively; P < 0.0001 for each). Moreover, of the patients who responded to pembrolizumab, the duration was more likely to be sustained at 3 years — 73% in the pembrolizumab 2 mg/kg arm and 74% in the pembrolizumab 10 mg/kg arm versus 13% in the chemotherapy arm [58]. Six-month PFS of 34% and 38% in the pembrolizumab 2 mg/kg and 10 mg/kg arms, respectively, was also significantly higher than with chemotherapy of 16% (P < 0.0001 for both). Although there were no significant differences in median OS (13.4/14.7 months for pembrolizumab 2 mg/kg and 10 mg/kg doses versus 11.0 months for chemotherapy), this was likely confounded by high crossover rates upon progression on chemotherapy (55%) [58]. Moreover, severe AEs were less frequent with pembrolizumab than chemotherapy (grade 3 10%/13% vs 20%; grade 4 0%/0% vs 6%) [57], and, importantly, health-related QoL was preserved to a greater degree for patients receiving pembrolizumab compared to chemotherapy (P = 0.01 for both doses of pembrolizumab compared with chemotherapy) [59].
Checkmate-037 compared nivolumab with investigator’s choice chemotherapy (dacarbazine or carboplatin–paclitaxel) in ipilimumab and BRAF/MEKi-refractory melanoma (1L +) [60]. Nivolumab resulted in superior ORR in this cohort (31.7% vs 10.6%) including increased duration of response; however, PFS and OS were not significantly improved (3.1 vs 3.4 months and 16 vs 14 months, respectively) [61•]. The study authors attributed this discrepancy to more patients with worse prognostic markers at baseline in the nivolumab cohort (including CNS metastases and elevated lactate dehydrogenase) and high treatment crossover rate in the chemotherapy group upon disease progression (41%).
Checkmate-066 compared nivolumab against dacarbazine in patients who had progressed on ipilimumab (1L +), highlighting significantly higher ORR (42.8% vs 14.4%, P < 0.001) [62]. Moreover, a landmark 5-year analysis demonstrated that only 2 patients (< 1%) experienced durable complete responses to dacarbazine, compared to 18% with nivolumab. This corresponded to significantly increased PFS and OS compared to dacarbazine, with 5-year PFS of 28% vs 3% and OS of 39% vs 17%, respectively. Importantly, nivolumab was also shown to maintain health-related QoL in these patients, unlike dacarbazine [63].
Fotemustine was compared with ipilimumab plus fotemustine and ipilimumab plus nivolumab in patients with asymptomatic brain metastasis (NIBIT-M2) in the 1L setting, showing vastly inferior response rates in both fotemustine-containing arms compared to ipilimumab–nivolumab (0% and 19.2% vs 44.4%, respectively) [64••]. In addition, OS at 4 years was significantly higher in the ipilimumab–nivolumab arm than the fotemustine and fotemustine–ipilimumab arms (41% vs 10.9% vs 10.3%, respectively).
Chemotherapy Versus Targeted Therapy
BRIM-3 was the randomised pivotal trial evaluating vemurafenib against dacarbazine in BRAF-mutant metastatic melanoma (1L), showing significant increased ORR (57% vs 9%, respectively; P < 0.001) and PFS (6.9 vs 1.6 months, respectively; P < 0.001) [65]. Subgroup analysis identified patients with favourable baseline factors such as higher performance status, normal LDH and reduced disease burden had greater benefit of vemurafenib compared to dacarbazine [66]. While a 4-year landmark analysis confirmed that this correlated to superior median OS in the vemurafenib groups (13.6 vs 9.7 months, respectively; P = 0.03), a similar majority of patients in both treatment arms had progressed (76.5 vs 74.4%, respectively) [66].
Similarly, dabrafenib was compared against dacarbazine (BREAK-3) in the 1L setting [67]. This study allowed patients to freely crossover to dabrafenib upon progression on dacarbazine, and thus, PFS was thus the primary endpoint to minimise the confounding effects on OS. ORR was superior in patients receiving first-line dabrafenib compared to dacarbazine (50% vs 6%, respectively) as was PFS (median 6.7 vs 2.9 months), which was consistent across subgroups stratified by performance status, LDH level and staging. However, durable responses were infrequent — with a 5-year PFS rate of only 12% in the dabrafenib arm compared to 0 in the dacarbazine, while the 5-year OS rate was similar between the two treatments (24% and 22%, respectively) [68]. BREAK-3 also provides valuable data on QoL benefits to patients — with a 6-week sub-analysis showing stable or improved QoL in patients receiving dabrafenib versus deterioration in those receiving dacarbazine [69].
Selumetinib is a MEK1/2 inhibitor with similar ORR in melanoma when compared to temozolomide (5.8 vs 9.4%, respectively) in the 1L setting, which was maintained in subgroup analysis of the BRAF-mutant cohort (11.1 vs 10.7%) [70]. Moreover, neither PFS nor OS were significantly extended by selumetinib compared to temozolomide (P = 0.70 and 0.10, respectively) — suggesting MEK inhibition is not superior to chemotherapy in unselected cohorts. In contrast, MEK inhibition with binimetinib in patients with NRAS mutations, present in 15–20% of cutaneous melanoma, shows significantly improved ORR when compared to dacarbazine in the 1L/1L + setting (15% vs 9%, respectively; P = 0.015) and PFS (2.8 vs 1.5 months, respectively; P < 0.001) [71]. Similarly, pimasertib was associated with significantly higher ORR (27% vs 14%, P = 0.05) and longer PFS (13 vs 7 months, P = 0.002) compared to dacarbazine (1L) [72]. However, neither study demonstrated significantly improved OS by MEK inhibition over chemotherapy (11.0 vs 10.1 months in NEMO, P = 0.50; 9 vs 11 months in NCT01693068).
METRIC is another randomised trial of trametinib versus investigators’ choice chemotherapy (dacarbazine or paclitaxel) in BRAF-mutant melanoma (1L/1L +) [73]. Higher ORR was observed in the trametinib versus chemotherapy arms (40% vs 14%, respectively); and, in addition, of the 65% of chemotherapy-treated patients who crossed over to trametinib, 44% then experienced an objective response. This corresponded to improved PFS (4.9 vs 1.5 months, respectively) as well as 5-year survival rates (32% vs 13.3%), even accounting for the high crossover rate. In addition, trametinib was associated with significantly reduced deterioration of QoL compared to chemotherapy over 12 weeks with less functional impairment and symptom burden [74].
When Can Chemotherapy Still Be Considered and When Should It Not?
All-in-all, given the lack of high-quality evidence supporting the meaningful benefits of chemotherapy in cutaneous melanoma, particularly given their inferiority to contemporary treatments and high toxicity rates, their utility is limited. However, there are a few scenarios in which chemotherapy may still be valuable. These are summarised in Table 3, including the evidence level supporting or discouraging their use (as per the Oxford Centre for Evidence-Based Medicine grading [75]).
Locoregional Chemotherapy
Approximately 5–10% of patients with melanoma will develop cutaneous in-transit metastases (ITMs) between the primary tumour and the draining nodal basin. Isolated limb perfusion/infusion involve regional administration of high-dose chemotherapy agents such as melphalan (an alkylating agent), with or without TNF-α, with pooled ORR of 60–80%, median OS of approximately 40 months and low rates of systemic toxicity in patients with ITM-limited disease [76, 77]. A phase 1b trial of isolated limb infusion in combination with nivolumab for ITMs is ongoing (NCT03685890). Electrochemotherapy with bleomycin (intravenous/intratumoural) can also be effective, with pooled ORR of 78% [78]. Given the paucity of data on contemporary treatments in patients with ITMs specifically, with one retrospective study demonstrating an ORR of 56% and 5-year OS of 63% with ICIs [79] (acknowledging likely differences between study cohorts), tumour-directed chemotherapeutics may be valuable alternatives to systemic therapy.
Salvage Therapy
Given that the majority of patients will ultimately progress on first-line treatments [2, 52], there remains a need to consider salvage treatment options for these patients. European Consensus guidelines suggest chemotherapy should only be considered as a last-line option in patients who have progressed on ICIs and (if BRAF-mutant) BRAF/MEKis, if enrolment in a clinical trial is not possible [80]. It has been postulated ICIs may potentiate efficacy of subsequent chemotherapy by priming CD8 + T cell activity, supported by small cohort studies [81]. However, a retrospective multicentre study demonstrated low ORR and PFS of 12.4% and 2.6 months, respectively, in patients treated with chemotherapy following ICI failure [82••]. Taxanes had the highest ORR (25%) and PFS (3.9 months); however, this was not statistically significant on multivariate analysis. This suggested overall low activity of chemotherapy which should not thus be the automatic treatment sequence following ICI/TT failure and emphasised the importance of instead seeking clinical trials or, if not feasible, informed patient-centred decision-making between chemotherapy versus BSC.
Similarly, in patients whom treatment with first-line therapies may be relatively contraindicated (e.g. ICIs in solid organ transplant recipients or those with uncontrolled autoimmune diseases [80]) who are thus also likely to be ineligible for clinical trials, the low clinical activity of chemotherapy should be carefully balanced with the potential harms to QoL when discussing active versus palliative therapy goals with patients. However, no trials specifically evaluated outcomes of chemotherapy in this population, and thus, high-quality evidence supporting or discouraging their use is lacking.
The role of predictive tumour biomarkers in guiding targeted chemotherapy regimens in melanoma remains largely unexplored. Alkylating agents such as dacarbazine, temozolomide and fotemustine induce DNA target lesions such as O6-methylguanine and O6-chlorethylguanine, which ordinarily can be repaired by O6-methylguanine-DNA methyltransferase (MGMT). Deficiency of MGMT may therefore indicate tumours more prone to alkylating chemotherapy, as is routinely screened in glioblastoma [83]. However, there are conflicting data correlating MGMT expression and response to dacarbazine between retrospective cohort analyses [84, 85], and no prospective studies investigate pre-treatment MGMT as a prognostic biomarker for chemotherapy in melanoma. Similarly, in vitro studies suggest mismatch repair protein deficiency (~ 25% of cutaneous melanoma) may predict resistance to alkylating agents in melanoma [86], while homologous recombination defects (~ 20% of cutaneous melanoma) may predict sensitivity to platins [87]. However, these have not been validated with clinical data. Pre-treatment biomarkers are an area in need of further research in melanoma to guide patient counselling and treatment stratification, particularly in the context of salvage therapy.
Brain Metastases
Most trials directly demonstrating inferiority of chemotherapy to ICIs and TT excluded patients with active brain metastases. Agents such as temozolomide or fotemustine may be considered in these patients given their reported intracranial ORRs of up to 25% [15]. Yet, both anti-CTLA-4- and anti-PD-1-based immunotherapies [88,89,90], as well as BRAF and MEK inhibitor combinations [91], have demonstrated higher ORRs and longer OS. Thus, chemotherapy should only be reserved for patients with CNS metastases who progress on or do not tolerate first-line ICIs/TT and trials are unavailable and always carefully considered against BSC.
Combined with Immunotherapy or Targeted Therapy
Prior to the introduction of ICIs, chemotherapy was frequently combined with early immunotherapies including IL-2 and IFN-α, although these combinations were more toxic and did not improve OS [51]. It has been suggested chemotherapy-induced necroptosis of cancer cells may increase immunogenic antigen exposure or upregulation of co-inhibitory ligands such as PD-L1, thereby augmenting ICI efficacy. Chemotherapy also has the potential to increase the ratio of effector to regulatory T cells in the tumour microenvironment to increase the cytotoxic potential of checkpoint inhibition, as observed in ex vivo models of docetaxel-treated breast cancer and cisplatin-treated lung cancer [92]. This corresponds to clinical trials showing greater efficacy of nab-paclitaxel and atezolizumab in triple-negative breast cancer [93] or pembrolizumab and platins in non–small cell lung cancer [94] compared to chemotherapy alone.
A small retrospective series of 60 melanoma patients who progressed on anti-PD-1 showed high activity of subsequent chemotherapy plus PD-1 inhibition (ORR 59% and median OS 3.5 years) [95•]. However, the prospective randomised trial NIBIT-M2 in patients with CNS metastases showed no meaningful benefits of ipilimumab addition to fotemustine chemotherapy, and in turn, both were vastly inferior to ipilimumab plus nivolumab [64••]. Moreover, 5-year survival of ipilimumab plus dacarbazine and ipilimumab alone is comparable (18 vs 26%, respectively) between the CA184-024 [62] and Checkmate-067 [3] trials (both in the 1L setting). While this warrants further research in melanoma, at present, there is limited evidence supporting addition of chemotherapy to ICIs, and these should not be used outside of clinical trials.
Similarly, pre-clinical data suggests chemotherapy plus TT may inhibit ATM-dependent DNA repair to kill melanoma cells, including in BRAF wild-type lines [96]. In vitro data also suggests a possible synergistic effect of sequencing temozolomide after vemurafenib in BRAF-mutant melanoma cells, without cross-resistance [97]. One randomised trial suggests the addition of trametinib to paclitaxel improves ORR (42% vs 13%, P = 0.01) and PFS (5.2 vs 3.4 months, P = 0.04) but not OS (9.4 vs 10.8 months, P = 0.18) compared to paclitaxel monotherapy in BRAF wild-type melanoma, although this was not compared against trametinib alone [98]. Therefore, no evidence supports combined chemotherapy and TT at present beyond clinical trials.
Bridge to Treatment Availability
Many first-line recommended treatments for melanoma as per contemporary guidelines are associated with significant costs and may not be readily available to every patient. For example, a 2016 availability analysis identified that, other than dacarbazine, almost all lower-income European countries either did not have access to contemporary melanoma treatments or, if they did, patients were subjected to full out-of-pocket costs [99]. Furthermore, a 2020 survey of oncologists in 82 countries suggested only 9–54% of patients in low- to middle-resource countries would have universal availability to critical cancer treatments, particularly ICIs [100]. In such cases, chemotherapy may be considered to reduce melanoma-related symptoms in patients with high tumour burden (particularly regimens with consistent ORRs and manageable AEs such as dacarbazine, fotemustine or nab-paclitaxel) even if survival is not significantly impacted while awaiting access.
Similarly, chemotherapy can be considered as a ‘bridge’ in selected cases while awaiting recruitment in a clinical trial. This can include preserving or reducing the deterioration of performance status which may affect otherwise elibility [101] or preserving biochemical markers such as LDH as baseline prognostic factors upon subsequent treatment with immunotherapy or TT [102].
Conclusion
Although chemotherapy has been widely studied and used in the historical treatment of cutaneous melanoma, its current utility is limited considering the significant superiority of ICIs and BRAF/MEKis. Chemotherapy agents are associated with low objective responses and unclear survival benefits compared to BSC, while their treatment-related toxicities may compromise patient QoL to a greater degree. Therefore, with a few exceptions such as locoregional tumour-directed chemotherapy, salvage therapy after first-line treatment failure (where clinical trials are not available) and as a bridge to treatment availability, traditional cytotoxic chemotherapy regimens should not be considered as standard-of-care in the contemporary management of metastatic melanoma. Our review has focused on metastatic cutaneous melanoma, and therefore, rarer subtypes such as mucosal or uveal melanoma, with their associated evolving treatment paradigms, are beyond the scope of this report.
Data Availability
Data sharing not applicable as no new data were generated in this manuscript.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gogas HJ, Kirkwood JM, Sondak VK. Chemotherapy for metastatic melanoma: time for a change? Cancer: Interdiscip Int J Am Cancer Soc. 2007;109:455–64.
Hamid O, Robert C, Daud A, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 2019;30:582–8.
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381:1535–46.
Seth R, Messersmith H, Kaur V, et al. Systemic therapy for melanoma: ASCO guideline. J Clin Oncol. 2020;38:3947–70.
Coit DG, Thompson JA, Albertini MR, et al. Cutaneous melanoma, version 2.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2019;17:367–402.
Keilholz U, Ascierto PA, Dummer R, et al. ESMO consensus conference recommendations on the management of metastatic melanoma: under the auspices of the ESMO Guidelines Committee. Ann Oncol. 2020;31:1435–48.
Ascierto PA, Dréno B, Larkin J, et al. 5-year outcomes with cobimetinib plus vemurafenib in BRAFV600 mutation–positive advanced melanoma: extended follow-up of the coBRIM study. Clin Cancer Res. 2021;27:5225–35.
Dummer R, Flaherty KT, Robert C, et al. COLUMBUS 5-year update: a randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600–mutant melanoma. J Clin Oncol 2022:JCO.21.02659.
Robert C, Grob JJ, Stroyakovskiy D, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N Engl J Med. 2019;381:626–36.
Crosby T, Fish R, Coles B, Mason MD. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst Rev. 2000:CD001215.
Huncharek M, Caubet JF, McGarry R0. Single-agent DTIC versus combination chemotherapy with or without immunotherapy in metastatic melanoma: a meta-analysis of 3273 patients from 20 randomized trials. Melanoma Res. 2001;11:75–81.
Quirt I, Verma S, Petrella T, Bak K, Charette M. Temozolomide for the treatment of metastatic melanoma: a systematic review. Oncologist. 2007;12:1114–23.
Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18:158-.
Avril MF, Aamdal S, Grob JJ, et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol. 2004;22:1118–25.
Jacquillat C, Khayat D, Banzet P, et al. Final report of the French multicenter phase II study of the nitrosourea fotemustine in 153 evaluable patients with disseminated malignant melanoma including patients with cerebral metastases. Cancer. 1990;66:1873–8.
Wiernik PH, Einzig AI. Taxol in malignant melanoma. J Natl Cancer Inst Monogr. 1993;15:185–7.
Einzig AI, Hochster H, Wiernik PH, et al. A phase II study of taxol in patients with malignant melanoma. Invest New Drugs. 1991;9:59–64.
Legha SS, Ring S, Papadopoulos N, Raber M, Benjamin RS. A phase II trial of taxol in metastatic melanoma. Cancer. 1990;65:2478–81.
O’Day S, Gonzalez R, Lawson D, et al. Phase II, randomized, controlled, double-blinded trial of weekly elesclomol plus paclitaxel versus paclitaxel alone for stage IV metastatic melanoma. J Clin Oncol. 2009;27:5452–8.
Bedikian AY, Plager C, Papadopoulos N, Eton O, Ellerhorst J, Smith T. Phase II evaluation of paclitaxel by short intravenous infusion in metastatic melanoma. Melanoma Res. 2004;14:63–6.
Walker L, Schalch H, King DM, Dietrich L, Eastman M, Kwak M, Kim K, Albertini MR. Phase II trial of weekly paclitaxel in patients with advanced melanoma. Melanoma Res. 2005;15:453–9.
Hersh EM, Del Vecchio M, Brown MP, et al. A randomized, controlled phase III trial of nab-paclitaxel versus dacarbazine in chemotherapy-naïve patients with metastatic melanoma. Ann Oncol. 2015;26:2267–74.
Bedikian AY, DeConti RC, Conry R, et al. Phase 3 study of docosahexaenoic acid–paclitaxel versus dacarbazine in patients with metastatic malignant melanoma. Ann Oncol. 2011;22:787–93.
Bedikian AY, Weiss GR, Legha SS, et al. Phase II trial of docetaxel in patients with advanced cutaneous malignant melanoma previously untreated with chemotherapy. J Clin Oncol. 1995;13:2895–9.
Einzig AI, Schuchter LM, Recio A, Coatsworth S, Rodriquez R, Wiernik PH. Phase II trial of docetaxel (Taxotere) in patients with metastatic melanoma previously untreated with cytotoxic chemotherapy. Med Oncol. 1996;13:111–7.
Aamdal S, Wolff I, Kaplan S, et al. Docetaxel (Taxotere) in advanced malignant melanoma: a phase II study of the EORTC Early Clinical Trials Group. Eur J Cancer. 1994;30:1061–4.
Kirkwood JM, Agarwala SS. Systemic cytotoxic and biologic therapy of melanoma. PPO Updates. 1993;7:1993–2016.
Glover D, Ibrahim J, Kirkwood J, et al. Phase II randomized trial of cisplatin and WR-2721 versus cisplatin alone for metastatic melanoma: an Eastern Cooperative Oncology Group Study (E1686). Melanoma Res. 2003;13:619–26.
Evans LM, Casper ES, Rosenbluth R. Phase II trial of carboplatin in advanced malignant melanoma 1, 2. Cancer Treat Rep. 1987;71:171.
Güven K, Kittler H, Wolff K, Pehamberger H. Cisplatin and carboplatin combination as second-line chemotherapy in dacarbazine-resistant melanoma patients. Melanoma Res. 2001;11:411–5.
Rao RD, Holtan SG, Ingle JN, et al. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. Cancer: Interdiscip Int J Am Cancer Soc. 2006;106:375–82.
Hodi FS, Soiffer RJ, Clark J, Finkelstein DM, Haluska FG. Phase II study of paclitaxel and carboplatin for malignant melanoma. Am J Clin Oncol. 2002;25:283–6.
Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol. 2013;31:373.
Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17:2745-.
Hofmann MA, Hauschild A, Mohr P, Garbe C, Weichenthal M, Trefzer U, Drecoll U, Tilgen W, Schadendorf D, Kaatz M, Ulrich J. Prospective evaluation of supportive care with or without CVD chemotherapy as a second-line treatment in advanced melanoma by patient’s choice: a multicentre Dermatologic Cooperative Oncology Group trial. Melanoma Res. 2011;21:516–23.
Seigler HF, Lucas VS Jr, Pickett NJ, Huang AT. DTIC, CCNU, bleomycin and vincristine (BOLD) in metastatic melanoma. Cancer. 1980;46:2346–8.
Vuoristo M-S, Hahka-Kemppinen M, Parvinen L-M, et al. Randomized trial of dacarbazine versus bleomycin, vincristine, lomustine and dacarbazine (BOLD) chemotherapy combined with natural or recombinant interferon-α in patients with advanced melanoma. Melanoma Res. 2005;15:291–6.
Eggermont AMM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40:1825–36.
Wilson MA, Schuchter LM. Chemotherapy for melanoma. In: Kaufman HL, Mehnert JM, editors. Melanoma. Cham: Springer International Publishing; 2016. p. 209–29.
Bajetta E, Del Vecchio M, Bernard-Marty C, et al. Metastatic melanoma: chemotherapy. Semin Oncol. 2002;29:427–45.
Agarwala SS, Kirkwood JM, Gore M, et al. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: a phase II study. J Clin Oncol. 2004;22:2101–7.
Paul MJ, Summers Y, Calvert AH, et al. Effect of temozolomide on central nervous system relapse in patients with advanced melanoma. Melanoma Res. 2002;12.
Silverman RB, Holladay MW. The organic chemistry of drug design and drug action. Academic press. 2014.
Long HJ. Paclitaxel (Taxol): a novel anticancer chemotherapeutic drug. Mayo Clin Proc. 1994;69:341–5.
Weiss RB, Donehower RC, Wiernik PH, et al. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–8.
Yamamoto Y, Kawano I, Iwase H. Nab-paclitaxel for the treatment of breast cancer: efficacy, safety, and approval. Onco Targets Ther. 2011;4:123.
Bradley MO, Swindell CS, Anthony FH, et al. Tumor targeting by conjugation of DHA to paclitaxel. J Control Release. 2001;74:233–6.
Verweij J, Clavel M, Chevalier B. Paclitaxel (TaxolTM) and docetaxel (TaxotereTM): not simply two of a kind. Ann Oncol. 1994;5:495–505.
Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Legha SS, Ring S, Papadopoulos N, Plager C, Chawla S, Benjamin R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer. 1989;64:2024–9.
Pasquali S, Hadjinicolaou AV, Sileni VC, Rossi CR, Mocellin S. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst Rev. 2018:CD011123.
Ugurel S, Röhmel J, Ascierto PA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies. Eur J Cancer. 2016;53:125–34.
• Hanna TP, Nguyen P, Baetz T, Booth CM, Eisenhauer E. A population-based study of survival impact of new targeted and immune-based therapies for metastatic or unresectable melanoma. Clin Oncol. 2018;30:609–17. Significant in highlighting overall trend towards improved survival following the advent of targeted and immunotherapies.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26.
Maio M, Grob JJ, Aamdal S, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33:1191–6.
Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616.
Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–18.
Hamid O, Puzanov I, Dummer R, et al. Final overall survival for KEYNOTE-002: pembrolizumab (pembro) versus investigator-choice chemotherapy (chemo) for ipilimumab (ipi)-refractory melanoma. Ann Oncol. 2016;27:vi379.
Schadendorf D, Dummer R, Hauschild A, et al. Health-related quality of life in the randomised KEYNOTE-002 study of pembrolizumab versus chemotherapy in patients with ipilimumab-refractory melanoma. Eur J Cancer. 2016;67:46–54.
Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84.
•• Larkin J, Minor D, D’Angelo S, et al. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: a randomized, controlled, open-label phase III trial. J Clin Oncol. 2018;36:383. Significant in showing superiority of nivolumab compared to chemotherapy in long-term follow-up.
Robert C, Long GV, Brady B, et al. Five-year outcomes with nivolumab in patients with wild-type BRAF advanced melanoma. J Clin Oncol. 2020;38:3937–46.
Long GV, Atkinson V, Ascierto PA, et al. Effect of nivolumab on health-related quality of life in patients with treatment-naïve advanced melanoma: results from the phase III CheckMate 066 study. Ann Oncol. 2016;27:1940–6.
•• Di Giacomo AM, Chiarion-Sileni V, Del Vecchio M, et al. Primary analysis and 4-year follow-up of the phase III NIBIT-M2 trial in melanoma patients with brain metastases. Clin Cancer Res. 2021;27:4737–45. Significant in showing long-term superiority of immunotherapy over chemotherapy (fotemustine) for treating melanoma brain metastases.
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–32.
Chapman PB, Robert C, Larkin J, et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: final overall survival results of the randomized BRIM-3 study. Ann Oncol. 2017;28:2581–7.
Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. The Lancet. 2012;380:358–65.
Hauschild A, Ascierto PA, Schadendorf D, et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib monotherapy: analysis from phase 2 and 3 clinical trials. Eur J Cancer. 2020;125:114–20.
Grob JJ, Amonkar MM, Martin-Algarra S, et al. Patient perception of the benefit of a BRAF inhibitor in metastatic melanoma: quality-of-life analyses of the BREAK-3 study comparing dabrafenib with dacarbazine. Ann Oncol. 2014;25:1428–36.
Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma selumetinib versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67.
Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435–45.
Lebbé C, Dutriaux C, Lesimple T, et al. Pimasertib versus dacarbazine in patients with unresectable NRAS-mutated cutaneous melanoma: phase II, randomized, controlled trial with crossover. Cancers. 2020;12:1727.
Robert C, Flaherty K, Nathan P, et al. Five-year outcomes from a phase 3 METRIC study in patients with BRAF V600 E/K–mutant advanced or metastatic melanoma. Eur J Cancer. 2019;109:61–9.
Schadendorf D, Amonkar MM, Milhem M, et al. Functional and symptom impact of trametinib versus chemotherapy in BRAF V600E advanced or metastatic melanoma: quality-of-life analyses of the METRIC study. Ann Oncol. 2014;25:700–6.
Group OLoEW. " The Oxford 2011 Levels of evidence." Oxford Centre for Evidence-Based Medicine. http://www.cebmnet/indexaspx?o=56532011. Accessed 20 Oct 2022.
Moreno-Ramirez D, Cruz-Merino L, Ferrandiz L, Villegas-Portero R, Nieto-Garcia A. Isolated limb perfusion for malignant melanoma: systematic review on effectiveness and safety. Oncologist. 2010;15:416–27.
Read T, Lonne M, Sparks DS, et al. A systematic review and meta-analysis of locoregional treatments for in-transit melanoma. J Surg Oncol. 2019;119:887–96.
Petrelli F, Ghidini A, Simioni A, Campana LG. Impact of electrochemotherapy in metastatic cutaneous melanoma: a contemporary systematic review and meta-analysis. Acta Oncol. 2022;61:533–44.
Holmberg C-J, Ny L, Hieken TJ, et al. The efficacy of immune checkpoint blockade for melanoma in-transit with or without nodal metastases – a multicenter cohort study. Eur J Cancer. 2022;169:210–22.
Garbe C, Amaral T, Peris K, et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment - Update 2022. Eur J Cancer. 2022;170:256–84.
Bouchereau S, Chaplain L, Fort M, et al. Impact of prior treatment with immune checkpoint inhibitors on dacarbazine efficacy in metastatic melanoma. Br J Cancer. 2021;125:948–54.
•• Goldinger SM, Buder-Bakhaya K, Lo SN, et al. Chemotherapy after immune checkpoint inhibitor failure in metastatic melanoma: a retrospective multicentre analysis. Eur J Cancer. 2022;162:22–33. Significant in showing low clinical activity of chemotherapy as a salvage therapy following failure of first-line treatment options.
Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003.
Ma S, Egyhazi S, Martenhed G, Ringborg U, Hansson J. Analysis of O6-methylguanine-DNA methyltransferase in melanoma tumours in patients treated with dacarbazine-based chemotherapy. Melanoma Res. 2002;12:335–42.
Busch C, Geisler J, Lillehaug JR, Lønning PE. MGMT expression levels predict disease stabilisation, progression-free and overall survival in patients with advanced melanomas treated with DTIC. Eur J Cancer. 2010;46:2127–33.
Kubeček O, Kopecký J. Microsatellite instability in melanoma: a comprehensive review. Melanoma Res. 2016;26:545–50.
Kim KB, Soroceanu L, de Semir D, et al. Prevalence of homologous recombination pathway gene mutations in melanoma: rationale for a new targeted therapeutic approach. J Investig Dermatol. 2021;141:2028-36.e2.
Di Giacomo AM, Chiarion-Sileni V, Del Vecchio M, et al. Primary analysis and 4-year follow-up of the phase III NIBIT-M2 trial in melanoma patients with brain metastases. The NIBIT-M2 phase 3, multicenter, randomized clinical trial. Clin Cancer Res. 2021;27:4737–45.
Long GV, Atkinson V, Lo S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol. 2018;19:672–81.
Tawbi HA, Forsyth PA, Algazi A, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722–30.
Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863–73.
Roselli M, Cereda V, di Bari MG, et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology. 2013;2: e27025.
Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–21.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. 2018;378:2078–92.
• Vera Aguilera J, Paludo J, McWilliams RR, et al. Chemo-immunotherapy combination after PD-1 inhibitor failure improves clinical outcomes in metastatic melanoma patients. Melanoma Res. 2020;30:364–75. Significant in highlighting chemoimmunotherapy combining ICIs and chemotherapy as a potential avenue requiring further research in melanoma.
Alonso-Marañón J, Villanueva A, Piulats JM, et al. Combination of chemotherapy with BRAF inhibitors results in effective eradication of malignant melanoma by preventing ATM-dependent DNA repair. Oncogene. 2021;40:5042–8.
Roos WP, Quiros S, Krumm A, et al. B-Raf inhibitor vemurafenib in combination with temozolomide and fotemustine in the killing response of malignant melanoma cells. Oncotarget. 2014;5:12607.
Urbonas V, Schadendorf D, Zimmer L, et al. Paclitaxel with or without trametinib or pazopanib in advanced wild-type BRAF melanoma (PACMEL): a multicentre, open-label, randomised, controlled phase II trial. Ann Oncol. 2019;30:317–24.
Cherny N, Sullivan R, Torode J, Saar M, Eniu A. ESMO European Consortium Study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann Oncol. 2016;27:1423–43.
Fundytus A, Sengar M, Lombe D, et al. Access to cancer medicines deemed essential by oncologists in 82 countries: an international, cross-sectional survey. Lancet Oncol. 2021;22:1367–77.
Magnuson A, Bruinooge SS, Singh H, et al. Modernizing clinical trial eligibility criteria: recommendations of the ASCO-Friends of cancer research performance status work group broadened eligibility: performance status. Clin Cancer Res. 2021;27:2424–9.
Claps G, Faouzi S, Quidville V, Chehade F, Shen S, Vagner S, Robert C. The multiple roles of LDH in cancer. Nat Rev Clin Oncol. 2022;19:749–62.
Funding
Open access funding provided by University of Zurich
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ines P. da Silva had travel support by BMS and MSD and speaker fee by Roche, BMS and MSD. Reinhard Dummer has intermittent, project-focused consulting and/or advisory relationships with Novartis, Merck Sharp & Dohme (MSD), Bristol-Myers Squibb (BMS), Roche, Amgen, Takeda, Pierre Fabre, Sun Pharma, Sanofi, Catalym, Second Genome, Regeneron, Alligator, T3 Pharma, MaxiVAX SA, Pfizer and touchIME outside the submitted work. James P. Pham, Anthony M. Joshua and Simone M. Goldinger declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pham, J.P., Joshua, A.M., da Silva, I.P. et al. Chemotherapy in Cutaneous Melanoma: Is There Still a Role?. Curr Oncol Rep 25, 609–621 (2023). https://doi.org/10.1007/s11912-023-01385-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-023-01385-6