
Abstract
Purpose of Review
Autonomic neuropathies are a complex group of disorders and result in diverse clinical manifestations that affect the cardiovascular, gastrointestinal, urogenital, and sudomotor systems. We focus this review on the diagnosis and treatment of peripheral autonomic neuropathies. We summarize the diagnostic tools and current treatment options that will help the clinician care for individuals with peripheral autonomic neuropathies.
Recent Findings
Autonomic neuropathies occur often in conjunction with somatic neuropathies but they can also occur in isolation. The autonomic reflex screen is a validated tool to assess sympathetic postganglionic sudomotor, cardiovascular sympathetic noradrenergic, and cardiac parasympathetic (i.e., cardiovagal) function. Initial laboratory evaluation for autonomic neuropathies includes fasting glucose or oral glucose tolerance test, thyroid function tests, kidney function tests, vitamin-B12, serum, and urine protein electrophoresis with immunofixation. Other laboratory tests should be guided by the clinical context. Reduced intraepidermal nerve density on skin biopsy is a finding, not a diagnosis. Skin biopsy can be helpful in selected individuals for the diagnosis of disorders affecting small nerve fibers; however, we strongly discourage the use of skin biopsy without clinical–physiological correlation. Ambulatory blood pressure monitoring may lead to early identification of patients with cardiovascular autonomic neuropathy in the primary care setting. Disease-modifying therapies should be used when available in combination with nonpharmacological management and symptomatic pharmacologic therapies. Autonomic function testing can guide the therapeutic decisions and document improvement with treatment.
Summary
A systematic approach guided by the autonomic history and standardized autonomic function testing may help clinicians when identifying and/or counseling patients with autonomic neuropathies. Treatment should be individualized and disease-modifying therapies should be used when available.
Similar content being viewed by others


Introduction
Autonomic neuropathies are a group of disorders characterized by damage to small unmyelinated or thinly myelinated autonomic nerves that are responsible for the regulation of involuntary physiologic processes including, but not limited to, heart rate, blood pressure, respiration, digestion, and sexual function [1]. Peripheral autonomic neuropathies may result from diverse causes, such as inherited disorders, metabolic derangements, exposure to toxins or drugs, autoimmune or inflammatory diseases, or paraneoplastic disorders (Table 1). Autonomic neuropathies may occur in conjunction with somatic neuropathies, which can affect large sensory and/or motor fibers and/or small sensory fibers. Detailed characterization of the somatic nerve disorder (e.g., demyelinating vs. axonal injury) is helpful for diagnostic purposes. Autonomic neuropathies may also occur in isolation. Diabetes or metabolic syndrome are the most common causes of autonomic neuropathies. The prevalence of autonomic involvement in diabetes increases with the duration of the disease, but autonomic dysfunction can be an early manifestation of the disease or caused by treatments (Table 1) [2]. Some patients with objective evidence of impairment of autonomic nerves on testing do not have an identifiable etiology, even after extensive workup.
Knowledge of the anatomic, neurochemical, and functional organization of the autonomic nervous system is critical for the diagnosis and treatment of autonomic neuropathies. The peripheral autonomic nervous system can be divided into the sympathetic, parasympathetic, and enteric divisions. Impairment of cholinergic postganglionic sympathetic fibers is frequent in autonomic neuropathies and these pathologic findings account for the loss of sweating, but compensatory hyperhidrosis can be seen. The impairment of heart rate control (heart rate variability) is predominately due to parasympathetic denervation and can be an early manifestation of autonomic neuropathy, especially in the setting of diabetes and amyloidosis. Neurogenic orthostatic hypotension is caused by damage or loss of small autonomic fibers in afferent and efferent nerves of the baroreflex arcs, with impairment of sympathetic noradrenergic reflex vasoconstriction. When the splanchnic vascular bed is extensively denervated, orthostatic hypotension can also be marked. Disturbance of bladder function, erectile dysfunction, and pupillary abnormalities may also be the manifestations of peripheral nerve disease when autonomic fibers to these structures are involved.
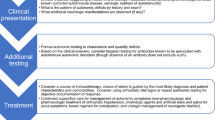
There have been recent reviews focusing on the clinical approach to patients with suspected autonomic neuropathies [3•, 4•]. The key part of the evaluation is a comprehensive autonomic history to define the temporal course of the symptoms and identify associated illnesses. The clinical evaluation and testing are guided by the history (Fig. 1) [5].

Management of cardiovascular autonomic neuropathy
Here, we review evidence-based recommendations for the diagnosis of autonomic neuropathies focusing on non-invasive laboratory evaluation of autonomic function, advances in the field of autoimmune and genetic autonomic disorders, and the role of tissue biopsy in the clinical setting. We also discuss the role of other non-invasive tests of autonomic function and the possible association between the coronavirus disease 2019 (COVID-19) and autonomic neuropathy. Finally, we review recent advances and future directions for the treatment of peripheral autonomic neuropathies focusing on disease-modifying therapies (when available) and symptomatic pharmacologic therapies.
Diagnosis of Autonomic Neuropathies: Evidence-Based Recommendations, Recent Advances, and Future Directions
Electrodiagnostic Assessment of the ANS—Autonomic Reflex Screen and Thermoregulatory Sweat Test
The goals of autonomic testing are to recognize the presence, distribution, and severity of autonomic dysfunction. In some cases, testing can detect patterns of autonomic impairment related to specific disorders. The American Autonomic Society, the International Society for Autonomic Neuroscience, the European Federation of Autonomic Societies, and the American Academy of Neurology have developed and endorsed consensus guidelines for clinical autonomic testing [6••]. The autonomic reflex screen is a validated approach to assess sympathetic postganglionic sudomotor, cardiovascular sympathetic noradrenergic, and cardiac parasympathetic (i.e., cardiovagal) function (Fig. 2) [6••]. Results should be compared to normative values for age and sex. The composite autonomic severity score (CASS), a validated instrument that quantifies the severity and distribution of autonomic failure, can be derived from the autonomic reflex screen [6••].

Illustrative example: autonomic reflex screen of a patient with diabetic autonomic neuropathy
Quantitative sudomotor axon reflex testing (QSART) assesses postganglionic sympathetic sudomotor function [7]. Cardiovagal function is assessed with heart rate responses to deep breathing (the most widely used method currently in clinical practice compared to the expiratory-inspiratory (E:I) ratio), the Valsalva ratio, and the vagal baroreflex sensitivity (slope of the relationship between RR interval and blood pressure in early phase II of the Valsalva maneuver) [8, 9]. Cardiovascular noradrenergic function is evaluated by assessing blood pressure responses to Valsalva maneuver and passive head-up tilt [8]. Quantitative indices of cardiovascular noradrenergic function, i.e., blood pressure recovery time or adrenergic baroreflex sensitivity, may be used [10]. Classic orthostatic hypotension is indicated by a sustained drop > 20 mmHg in systolic blood pressure and/or > 10 mmHg in diastolic blood pressure within 3 min during tilt table test. In delayed orthostatic hypotension, the drop in blood pressure becomes apparent after a prolonged standing posture (> 3 min). A neurogenic etiology of orthostatic hypotension is indicated by a lack of reflex vasoconstriction during the Valsalva maneuver [6••]. An insufficient compensatory increase in heart rate in relation to orthostatic hypotension is also a marker of neurogenic orthostatic hypotension [11••]. In a prospective study of 402 patients with orthostatic hypotension, a neurogenic etiology was reliably identified when the ratio of orthostatic heart rate changes over systolic blood pressure changes at 3 min of tilt was < 0.5 beats/min per mmHg [11••]. Importantly, similar findings were reported with active standing [12].
The integrity of central and peripheral sympathetic sudomotor pathways can be assessed by the thermoregulatory sweat test [13]. This test is performed in a temperature and humidity-controlled room and the body is warmed up to a core temperature of 38 °C. The patient lies supine with exposed body surface covered with an indicator powder that changes color when wet. Digital photographs with pixel counter quantify the percentage of anhidrosis [13].
Standardized autonomic function testing plays a role in accurate diagnosis of most autonomic neuropathies but also monitoring of treatment efficacy as demonstrated in autoimmune autonomic ganglionopathies [14••].
Although standardized autonomic function testing provides an accurate measure of autonomic function, one of the main limitations of electrodiagnostic assessment of the autonomic nervous system is the limited availability of dedicated autonomic laboratories and specialty trained clinicians.
Electrodiagnostic Assessment of the ANS—Other Tests of Autonomic Function
Additional autonomic tests may be used in select research settings but at this time are not practical for routine clinical use in patients with autonomic neuropathies. These tests include the study of sympathetic skin response, heart rate variability, corneal confocal microscopy, microneurography, electrical-evoked potentials, and laser-evoked potentials. The cold pressor test and analysis of the blood pressure response to sustained handgrip may have a role in patients with suspected involvement of the afferent limb of the baroreflex (e.g., afferent baroreflex failure) [15]. Quantitative sensory testing is a psychophysical assessment of different somatosensory thresholds and may be helpful in the evaluation of patients with small fiber neuropathy who often have both sensory and autonomic complaints; however, the results are highly dependent on methodology and the full cooperation of the subject [16].
Laboratory Tests
Initial laboratory evaluation for autonomic neuropathies includes complete blood count, basic metabolic panel, thyroid function tests, fasting glucose or oral glucose tolerance test, vitamin-B12, and serum and urine protein electrophoresis [4•]. Secondary causes of autonomic dysfunction should be ruled out and testing may include screening for adrenal insufficiency and pheochromocytoma. Other tests should be guided by clinical suspicion (e.g., IgA tissue transglutaminase antibodies if there is a suspicion of celiac disease).
Measurement of plasma catecholamines may be helpful in a few selected cases. For example, plasma norepinephrine is often elevated in patients with afferent baroreflex failure [17••].
Laboratory testing plays an important role in identifying autoimmune etiologies of autonomic neuropathy. If there is a clinical suspicion of autoimmunity, testing often includes antinuclear antibodies (ANA), extractable nuclear antigen antibodies (including SS-A/SS-B antibodies), C-reactive protein, and erythrocyte sedimentation rate. Some paraneoplastic nuclear antibodies are highly associated with autonomic neuropathies, especially the Type 1 Antineuronal Nuclear Autoantibody (ANNA-1 or anti-Hu) and Collapsing Response-Mediator Protein 5 antibody (CRMP-5 or Anti-CV2) [18]. The ɑ-3 nicotinic acetylcholine ganglionic antibodies are membrane receptor antibodies that are classically associated with autoimmune autonomic ganglionopathy. One should always interpret the presence of this antibody in light of the clinical presentation because they have also been reported in individuals without objective evidence of autonomic impairment. A level > 0.40 nmol/L had high specificity and moderate sensitivity for severe autonomic failure, whereas levels of less than 0.20 nmol/L may not be clinically relevant [19]. Karagiorgou and colleagues recently demonstrated that a cell-based assay for α3- nicotinic acetylcholine ganglionic antibodies is more specific for the diagnosis of autoimmune autonomic ganglionopathy compared to the commercially available radioimmunoassay precipitation assay [20•]. Further studies are necessary to investigate the clinical significance of trisulfated disaccharide IdoA2S-GlcNS-6S antibodies (TS-HDS), fibroblast growth factor receptor 3 (FGFR3), and muscarinic (M3) acetylcholine receptor antibodies in patients with autonomic neuropathies. Preliminary findings do not suggest a benefit from treatment with immune globulin in individuals with small fiber neuropathy with TS-HDS and FGFR3 autoantibodies [21].
Advances in genetic studies have led to the identification of new autonomic neuropathies and newly approved gene-modifying therapies. Hereditary transthyretin variant amyloidosis (ATTRv) is the most common form of hereditary amyloidosis. More than 130 mutations have been identified to date, and some mutations predominately cause polyneuropathy, whilst other mutations cause cardiomyopathy or mixed phenotypes [22]. Val30Met is the most common ATTRv mutation worldwide but the clinical presentation is variable among individuals with the mutation. Early suspicion and recognition of ATTRv amyloidosis can lead to an earlier diagnosis and treatment. If there is clinical suspicion of amyloid neuropathy, DNA sequencing of the transthyretin gene can support or exclude a diagnosis of ATTRv amyloidosis [23•]. Furthermore, genetic testing is helpful in healthy but potentially at-risk persons with a family history of ATTRv amyloidosis. However, the penetrance of ATTRv is incomplete in carriers. Therefore, the detection of amyloid deposits on biopsy samples is crucial to confirm the diagnosis [24••]. The sensitivity of biopsy in different sites varies but the specificity is high for a diagnosis of ATTRv amyloidosis [25]. Echocardiogram and cardiac magnetic resonance imaging are important investigations for detecting cardiac amyloidosis. In cases with hypertrophic cardiopathy with negative biopsy findings, radionuclide bone scintigraphy with technetium-labeled bisphosphonates can localize cardiac amyloid deposits and may obviate the need for endomyocardial biopsy [26]. Characterizing the subtype of amyloid protein by mass spectrometry or immunohistochemistry can enhance diagnostic accuracy and guide management [27, 28].
Autonomic dysfunction has been recently recognized in cerebellar ataxia, neuropathy, and vestibular areflexia—“CANVAS”—syndrome [29]. CANVAS is due to AAGGG repeat expansion in the second intron of replication factor C subunit 1 (RFC1) gene. In a case series of 23 patients, 83% had evidence of autonomic dysfunction; all patients had at least one autonomic symptom and 91% had more than two symptoms. Cold feet (78%), erectile dysfunction (78% of men), light-headedness (65%), constipation (65%), and sicca syndrome (52%) were the most common symptoms reported. Thirty percent of patients fulfilled the criteria for orthostatic hypotension. Autonomic dysfunction in CANVAS may be related to a primary ganglioneuropathy involving the autonomic, facial, trigeminal, vestibular, and sensory ganglia and their projections. Another study demonstrated that biallelic repeat expansions in RFC1 were a frequent cause of whole-exome sequencing negative hereditary sensory and autonomic neuropathy with chronic cough and ataxia [30]. Genetic testing with repeat-primed polymerase chain reaction to detect intronic expansions should be performed in cases with suspicion of CANVAS syndrome.
Skin Biopsy
Antibodies against the cytoplasmic protein gene product 9.5 (PGP9.5) can visualize the rich cutaneous innervation via bright-field immunohistochemistry. Evidence of reduced or absent intraepidermal nerve fibers on punch skin biopsy, a minimally invasive technique, may be used to support a diagnosis of small fiber neuropathy. The sensitivity and specificity of skin biopsy in the diagnosis of small fiber neuropathy vary between 78–92% and 65–90% respectively [31]. A diagnosis of small fiber neuropathy is made when nerve fiber density is in the lowest 5th percentile compared to normative values for both age and gender in the appropriate clinical context [32]. Punch skin biopsy is billable to insurance companies and many commercial laboratories are claiming to provide the same service and diagnostic accuracy as research laboratories. This has led to an increase in testing and diagnosis, often without clinical–physiological–pathological correlation.
Several points are worth discussing. First, reduced intraepidermal nerve density is a finding, not a diagnosis. Skin biopsy alone should not be used to diagnose small fiber neuropathy. PGP9.5 staining does not provide any information about small nerve fiber functions. This may explain the lack of correlation between intraepidermal nerve fiber density and neuropathic pain [33]. The use of other molecular markers linked to different subtypes of skin nerve fibers might provide a better clinical-physiological-pathological correlation [34]. Second, objective morphometric assessment of small nerve fibers should be performed in a body region where there are symptoms and clinicians should always correlate the biopsy findings with the clinical context taking into consideration that functional changes are not always associated with the loss of nerve fibers. For example, conditions associated with neuropathic pain secondary to some channelopathies are not always associated with a reduction in intraepidermal nerve fiber density [35]. Finally, the analysis of skin biopsy results should follow a rigorous process: (1) results of intraepidermal nerve fiber density should be interpreted based on the site where the biopsy is taken because control data exist only for a few body sites; (2) the results should be compared with a control population using published data; and (3) the report should mention standard deviations or interquartile intervals, and percentage of truly abnormal by a predefined outcome measure.
Skin biopsies may also be helpful to identify pathological hallmarks in different diseases. For example, visualization of amyloid deposition confirmed by Congo red stain in the subcutaneous tissue is sensitive and specific for diagnosing systemic amyloidosis [36]. Skin biopsy also has great potential for the diagnosis of synucleinopathies [37]. However, alpha-synuclein deposit in cutaneous autonomic nerve fibers has also been found in individuals with postural tachycardia with neuropathic features or long-COVID [38•, 39•]. Further studies are needed to confirm whether alpha-synuclein deposition in these individuals reflects a reactive process or an early stage of more widespread synucleinopathies.
Ambulatory Blood Pressure Monitoring—an Underutilized Tool in Autonomic Neuropathies?
Ambulatory blood pressure monitoring (ABPM) provides some advantages over the measurement of orthostatic vital signs in the autonomic laboratory or during office visits. It allows for “real-life” assessment while patients perform daily activities; it determines diurnal and nocturnal blood pressure patterns that are characteristics of autonomic failure such as supine hypertension, labile blood pressure, non-dipping of nocturnal blood pressure; it also allows for the evaluation of the impact of antihypertensive and pressor medications [40]. One study reported that daytime blood pressure variability was a good screening tool to identify patients with autonomic failure from controls; however, correlation analysis between quantitative assessment of autonomic function and ambulatory blood pressure was not performed [41]. There is a need to standardize parameters derived from ABPM. High variability of blood pressure with low heart rate variability over a 24 h may allow for accurate identification of patients with autonomic neuropathy with autonomic failure. This approach may lead to early identification of patients with autonomic neuropathy in the primary care setting, earlier referral to specialized autonomic evaluation if necessary, reduce delay in treatment, and decrease unnecessary testing.
COVID-19 and Autonomic Neuropathy—What Is the Evidence?
The novel severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) infection has been associated with para-infectious neurological and autonomic abnormalities [42••]. There is concern that post-acute sequelae of COVID-19 (also called “long-haul COVID” “long-COVID” or “post-COVID syndrome”) include abnormalities of autonomic cardiovascular function, which may be associated with various symptoms such as fatigue, “brain fog,” sensory changes, and orthostatic intolerance [43]. The pathophysiological mechanisms of post-acute cardiovascular autonomic dysfunction in COVID-19 are not well understood. Proposed mechanisms include dysfunction of the extended autonomic nervous system, immune dysregulation triggered by the infection, dysfunction of the renin–angiotensin–aldosterone system, or a restricted autonomic neuropathy causing impaired vasomotor and venomotor tone in the lower limbs among others [44]. Cases of autonomic dysfunction beyond postural tachycardia syndrome have been reported, including phenomena such as small fiber neuropathy [45], neurocardiogenic syncope, and post-COVID-19 exacerbation of paroxysmal hypothermia and hyperhidrosis [46]. Post-COVID-19 acute inflammatory demyelinating polyneuropathy has been described as well [47]. In our experience, however, most individuals with long-COVID who present to the autonomic clinic do not have objective evidence of autonomic neuropathy. In a retrospective series of 27 patients with a confirmed history of COVID-19 infection referred for autonomic testing for symptoms concerning for para-/postinfectious autonomic dysfunction, autonomic symptoms developed between 0 and 122 days following the acute infection and included lightheadedness (93%), orthostatic headache (22%), syncope (11%), hyperhidrosis (11%), and burning pain (11%). Twenty-two percent of patients fulfilled the criteria for postural tachycardia syndrome, and 11% had borderline findings to support orthostatic intolerance. The most common autonomic testing finding was orthostatic intolerance, often without objective hemodynamic abnormalities on testing [48••]. In a study of 16 young adults who tested positive for SARS-CoV-2, resting heart rate and blood pressure were not different compared to controls (N = 14); however, muscle sympathetic nerve activity was higher in COVID subjects compared to controls during head-up tilt with a group-by-position interaction in muscle sympathetic nerve activity burst incidence, as well as heart rate, in response to tilt. The results of this study suggest that These results indicate resting sympathetic activity may be elevated following SARS-CoV-2 infection [49]. Further studies are necessary to investigate the association between autonomic neuropathy and COVID-19.
Treatment of Autonomic Neuropathies
The treatment of autonomic neuropathies is based on the combination of disease-modifying therapies (when available), nonpharmacological management, and symptomatic pharmacologic therapies. The autonomic reflex screen is helpful to identify initial deficits and guide treatment [14••].
Disease-Modifying Therapies—Focus on Hereditary Transthyretin Amyloidosis
Liver transplantation replaces the primary source of mutant transthyretin. Liver transplantation is associated with improved long-term survival in hereditary transthyretin amyloidosis. Nevertheless, peripheral neuropathy and cardiomyopathy may progress following liver transplantation and genotype influences post-transplantation survival [50]. Furthermore, adverse effects of surgery and complications of long-term immunosuppression are limitations of this approach. Novel treatments for hereditary transthyretin amyloidosis (hATTR) are now available. Tafamidis meglumine, a kinetic TTR stabilizer, can be administered orally and prevents misfolding and deposition of mutated TTR [51]. Two studies have demonstrated some benefit of tafamidis in early-stage hATTR polyneuropathy compared to placebo [52, 53]. Outcomes were mixed in patients with mid-and late-stage hATTR and worsening of baseline autonomic function and cardiac disease has been reported [54]. Diflunisal, a nonsteroidal anti-inflammatory drug, has TTR-stabilizing properties; however, adverse events such as renal failure or thrombocytopenia may limit its use. Oligonucleotide-based gene therapies (patisiran and inotersen) have been recently approved by the Food and Drug Administration in the United States for the treatment of hATTR. These agents have demonstrated efficacy in patients with early-and late-stage disease and improve, halt, or slow neuropathy progression.
Symptomatic Pharmacologic Therapies—Focus on Cardiovascular Manifestations of Autonomic Neuropathies
Neurogenic Orthostatic Hypotension
Orthostatic hypotension is associated with a risk of falls, cognitive impairment, and dementia [55]. The nonpharmacological management of orthostatic hypotension should be individualized (Table 2) [56••]. Available options for the pharmacological management of neurogenic orthostatic hypotension are limited. Fludrocortisone is a synthetic mineralocorticosteroid that can be helpful in combination with adequate hydration and liberalizing dietary sodium. Side effects of fludrocortisone include supine hypertension and hypokalemia [57]. Droxidopa (a precursor of norepinephrine) and midodrine (an alpha-receptor agonist) are the only drugs approved by the Food and Drug Administration for the treatment of neurogenic orthostatic hypotension [57]. Droxidopa is most beneficial in patients with low supine plasma norepinephrine levels (< 200 pg/mL) [58]. Pyridostigmine, a reversible inhibitor of acetylcholinesterase, increases the availability of acetylcholine to bind to muscarinic or nicotinic receptors. The pressor effect of pyridostigmine may be suboptimal compared to other drugs, but it can be used as an add-on therapy in patients with moderate to severe autonomic failure [59].
Norepinephrine transporter (NET) inhibition is a mechanism that is being studied to increase blood pressure in patients with autonomic failure. Shibao and colleagues first showed that atomoxetine induced a hypertensive response in a small group of patients with central autonomic failure [60,61,62]. In a recent study, Kaufmann and colleagues reported the safety and efficacy of ampreloxetine to treat neurogenic orthostatic hypotension, in a small phase II clinical trial [63•]. This study demonstrated that Ampreloxetine was safe and improved orthostatic symptoms and seated/standing blood pressure with little change in supine blood pressure [63•]. Unfortunately, a press release from the company announced the failure of the phase III study due to a lack of efficacy [64]. We are awaiting for the results of another phase II trial that is investigating the effect of atomoxetine in the symptomatic management of nOH (ClinicalTrials.gov: NCT02796209).
Most patients with orthostatic hypotension and supine hypertension have normal seated blood pressures. Therefore, it is important to consider the 24-h blood pressure profile and not to use the sitting blood pressure as a target to assess treatment efficacy. Although it is crucial to avoid iatrogenic causes of orthostatic hypotension, there is no clear epidemiological relationship between orthostatic hypotension and the use of antihypertensive medications. A recent meta-analysis of 18,466 individuals from randomized clinical trials for the treatment of essential hypertension has shown that blood pressure-lowering treatment decreased the risk for orthostatic hypotension [65••]. Because individuals with autonomic neuropathy are at increased risk of cardiac arrhythmia and sudden death [66], it is important to avoid the use of medications that affect cardiac repolarization and consequently prolong the QT if possible.
Supine Hypertension
Supine hypertension is defined as a systolic blood pressure of at least 140 mmHg and/or diastolic blood pressure of at least 90 mmHg [67]. More than 50% of patients with neurogenic orthostatic hypotension may suffer from supine hypertension [68]. Treatment of supine hypertension can worsen OH and vice versa. Nocturnal pressure natriuresis from supine hypertension also worsens orthostatic hypotension. Supine hypertension is associated with cardiac hypertrophy and kidney failure, which may increase cardiovascular and renal disease, and mortality. Supine hypertension is also linked to impaired cerebral autoregulation [69••]. Short-acting antihypertensives such as angiotensin II receptor blockers (e.g., low dose losartan) and calcium channel blockers (e.g., nifedipine) might be considered (Table 2). On the other hand, alpha-blockers and diuretics can exacerbate orthostatic hypotension and should be avoided. Some patients with supine hypertension retain the normal pattern of blood pressure dipping at night, with elevated blood pressure mostly early in the night. These patients may benefit from ingestion of a carbohydrate-rich snack at bedtime to induce postprandial hypotension [70]. In one study, local heat therapy also effectively lowered overnight blood pressure in patients with autonomic failure and supine hypertension [71•].
Labile Blood Pressure
It is recommended not to “chase” the blood pressure in cases with blood pressure lability secondary to afferent baroreflex failure. Central sympatholytic drugs such as clonidine can be helpful [72]. Vasodilators should be used with caution because they can elicit profound hypotension. Patients should be aware that medications that modify the vascular tone or sympathetic activity can have a drastic effect on the blood pressure precipitating hypertensive crisis or hypotension. Peripheral decarboxylase inhibition with carbidopa reduces blood pressure variability in patients with familial dysautonomia who have afferent lesions of the arterial baroreflex [73]. Further studies are necessary to test this approach in individuals with acquired afferent baroreflex failure.
Tachycardia
Symptomatic manifestations of cardiac autonomic neuropathy may include resting sinus tachycardia and exercise intolerance. Higher resting heart rate (> 78 bpm) and a rise in heart rate with time are independent risk predictors for all-cause mortality in patients with diabetes [74]. A fixed heart rate that is unresponsive or poorly responsive (i.e., chronotropic incompetence) to exercise indicates almost complete cardiac denervation. After exclusion of other causes of tachycardia (e.g., anemia, medication, cardiac arrhythmia, recreational drugs), the use of β-adrenergic blockers may be indicated.
Multidisciplinary Care
A multidisciplinary approach involving different specialties is necessary for optimal management of the diverse clinical manifestations of autonomic neuropathies.
Conclusion
A detailed history and neurological examination guide testing in individuals with peripheral autonomic neuropathies. Workup of autonomic neuropathies may include standardized autonomic function testing, laboratory testing, imaging, and tissue biopsy. Treatment should be individualized and disease-modifying therapies should be used when available. Further studies are necessary to investigate the association between autonomic neuropathy and COVID-19.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gibbons CH. Basics of autonomic nervous system function. Handb Clin Neurol. 2019;160:407–18.
Freeman R. Diabetic autonomic neuropathy. Handb Clin Neurol. 2014;126:63–79.
• Kaur D, Tiwana H, Stino A, Sandroni P. Autonomic neuropathies. Muscle Nerve. 2021;63(1):10–21. This is an excellent review which categorizes peripheral autonomic neuropathies based on their acute/subacute or chronic onset.
• Lamotte G, Sandroni P. A practical approach to peripheral autonomic neuropathies. Curr Opin Neurol. 2021;34(5):638–47. This review focuses on the practical approach of individuals with peripheral autonomic neuropathies with an emphasis on the autonomic history and physical examination.
Goldstein DS, Cheshire WP Jr. The autonomic medical history. Clin Auton Res. 2017;27(4):223–33.
•• Cheshire WP, Freeman R, Gibbons CH, et al. Electrodiagnostic assessment of the autonomic nervous system: a consensus statement endorsed by the American Autonomic Society, American Academy of Neurology, and the International Federation of Clinical Neurophysiology. Clin Neurophysiol. 2021;132(2):666–82. This is the most recent consensus statement on autonomic testing.
Low PA, Caskey PE, Tuck RR, Fealey RD, Dyck PJ. Quantitative sudomotor axon reflex test in normal and neuropathic subjects. Ann Neurol. 1983;14:573–80.
Low PA. Autonomic nervous system function. J Clin Neurophysiol. 1993;10(1):14–27.
Pickering TG, Sleight P. Quantitative index of baroreflex activity in normal and hypertensive subjects using Valsalva’s manoeuvre. Br Heart J. 1969;31(3):392.
Vogel ER, Sandroni P, Low PA. Blood pressure recovery from Valsalva maneuver in patients with autonomic failure. Neurology. 2005;65:1533–7.
•• Norcliffe-Kaufmann L, Kaufmann H, Palma JA, et al. Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies. Ann Neurol. 2018;83(3):522–31. This important study validated the ratio of orthostatic heart rate changes over systolic blood pressure changes to identify individuals with neurogenic orthostatic hypotension.
Balagny P, Wanono R, d’Ortho MP, Vidal-Petiot E. Reply to validation of the new diagnostic tests for neurogenic orthostatic hypotension. Ann Neurol. 2018;84(6):957–8.
Fealey RD, Low PA, Thomas JE. Thermoregulatory sweating abnormalities in diabetes mellitus. Mayo Clin Proc. 1989;64(6):617–28.
•• Koay S, Vichayanrat E, Bremner F, et al. Multimodal biomarkers quantify recovery in autoimmune autonomic ganglionopathy. Ann Neurol. 2021;89(4):753–68. This study demonstrated the role of quantitative autonomic function testing to monitor the response to treatment in individuals with autoimmune autonomic ganglionopathy.
Robertson D, Hollister AS, Biaggioni I. Arterial baroreflex failure in man. Clin Auton Res. 1993;3:212.
Potockova V, Mala S, Hoskovcova L, et al. Thermal quantitative sensory testing as a screening tool for cardiac autonomic neuropathy in patients with diabetes mellitus. Brain Behav. 2022;12(3): e2506.
•• Lamotte G, Coon EA, Suarez MD, et al. Standardized autonomic testing in patients with probable radiation-induced afferent baroreflex failure. Hypertension. 2022;79(1):50–6. This study described the pattern of autonomic dysfunction on standardized autonomic testing in patients with probable radiation-induced afferent baroreflex failure.
Shouman K, Sandroni P. Antibody testing for suspected autoimmune autonomic dysfunction and small fiber neuropathies. J Clin Neurophysiol. 2021;38(4):274–8.
Cutsforth-Gregory JK, McKeon A, Coon EA, et al. Ganglionic antibody level as a predictor of severity of autonomic failure. Mayo Clin Proc. 2018;93(10):1440–7.
• Karagiorgou K, Dandoulaki M, Mantegazza R, et al. Novel cell-based assay for alpha-3 nicotinic receptor antibodies detects antibodies exclusively in autoimmune autonomic ganglionopathy. Neurol Neuroimmunol Neuroinflamm. 2022;9(3):e1162. https://doi.org/10.1212/NXI.0000000000001162. Print 2022 May.
Gibbons CH. A double-blind, placebo controlled trial of Gamunex-C in patient with small fiber neuropathy associated with autoantibodies to TS-HDS and FGFR3 (P8-13.002). Neurology. 2022;98(18 Supplement).
Gertz MA, Mauermann ML, Grogan M, Coelho T. Advances in the treatment of hereditary transthyretin amyloidosis: a review. Brain Behav. 2019;9(9): e01371.
• Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–22. This review outlines detailed recommendations to improve the diagnosis of ATTR amyloidosis with peripheral neuropathy.
•• Carroll A, Dyck PJ, de Carvalho M, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol, Neurosurg Psychiatr 2022. This review highlights the key advances in the diagnostic techniques, current and emerging management strategies, and biomarker development for disease progression in hereditary transthyretin amyloidosis.
Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Current opinion in neurology. 2016;29(Suppl 1):S14–26.
Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12.
Klein CJ, Vrana JA, Theis JD, et al. Mass spectrometric-based proteomic analysis of amyloid neuropathy type in nerve tissue. Arch Neurol. 2011;68(2):195–9.
Rezk T, Gilbertson JA, Mangione PP, et al. The complementary role of histology and proteomics for diagnosis and typing of systemic amyloidosis. J Pathol Clin Res. 2019;5(3):145–53.
Wu TY, Taylor JM, Kilfoyle DH, et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia “CANVAS” syndrome. Brain. 2014;137(Pt 10):2649–56.
Beijer D, Dohrn MF, De Winter J, et al. RFC1 repeat expansions: a recurrent cause of sensory and autonomic neuropathy with cough and ataxia. Eur J Neurol. 2022;29(7):2156–61.
Hovaguimian A, Gibbons CH. Diagnosis and treatment of pain in small-fiber neuropathy. Curr Pain Headache Rep. 2011;15(3):193–200.
Provitera V, Gibbons CH, Wendelschafer-Crabb G, et al. A multi-center, multinational age- and gender-adjusted normative dataset for immunofluorescent intraepidermal nerve fiber density at the distal leg. Eur J Neurol. 2016;23(2):333–8.
Shun CT, Chang YC, Wu HP, et al. Skin denervation in type 2 diabetes: correlations with diabetic duration and functional impairments. Brain. 2004;127(Pt 7):1593–605.
Baskozos G, Sandy-Hindmarch O, Clark AJ, et al. Molecular and cellular correlates of human nerve regeneration: ADCYAP1/PACAP enhance nerve outgrowth. Brain. 2020;143(7):2009–26.
Bennett DL, Woods CG. Painful and painless channelopathies. The Lancet Neurology. 2014;13(6):587–99.
Wu B, Pak DM, Smith KD, Shinohara MM. Utility of abdominal skin punch biopsy for detecting systemic amyloidosis. J Cutan Pathol. 2021;48(11):1342–6.
Wang N, Gibbons CH, Lafo J, Freeman R. alpha-Synuclein in cutaneous autonomic nerves. Neurology. 2013;81(18):1604–10.
• Levine TD, Bellaire B, Gibbons C, Freeman R. Cutaneous alpha-synuclein deposition in postural tachycardia patients. Ann Clin Transl Neurol. 2021;8(4):908–17. In this study, a subgroup of patients with postural tachycardia had evidence of cutaneous alpha-synuclein deposition.
• Miglis MG, Seliger J, Shaik R, Gibbons CH. A case series of cutaneous phosphorylated α-synuclein in Long-COVID POTS. Clin Auton Res: Off J Clin Auton Res Soc. 2022;32(3):209–12. In this research letter, the authors report phosphorylated alpha-synuclein in skin biopsy samples in five young patients with long-COVID postural tachycardia syndrome.
Guidelines for the clinical use of 24 hour ambulatory blood pressure monitoring (ABPM) (JCS 2010): digest vesrion. Circ J. 2012;76(2):508–19.
Lodhi HA, Peri-Okonny PA, Schesing K, et al. Usefulness of blood pressure variability indices derived from 24-hour ambulatory blood pressure monitoring in detecting autonomic failure. J Am Heart Assoc. 2019;8(7): e010161.
•• Spudich S, Nath A. Nervous system consequences of COVID-19. Science. 2022;375(6578):267–9. This is an excellent review of the neurological complications of COVID-19.
Goldstein DS. Principles of Autonomic Medicine. https://neuroscience.nih.gov/publications/Principles%20of%20Autonomic%20Medicine%20v.%202.1.pdf2020. Accessed 1 Nov 2022.
Goldstein DS. The extended autonomic system, dyshomeostasis, and COVID-19. Clin Auton Res: Off J Clin Auton Res Soc. 2020;30(4):299–315.
Novak P. Post COVID-19 syndrome associated with orthostatic cerebral hypoperfusion syndrome, small fiber neuropathy and benefit of immunotherapy: a case report. eNeurologicalSci. 2020;21:100276.
Lamotte G, Benarroch EE, Coon EA. Paroxysmal hypothermia and hyperhidrosis with exacerbation after COVID-19 Infection. Clin Auton Res: Off J Clin Auton Res Soc. 2021;31(2):327–9.
Mostel Z, Ayat P, Capric V, Trimmingham A, McFarlane SI. Guillain-Barré syndrome in a COVID-19 patient: a case report and review of management strategies. Am J Med Case Rep. 2021;9(3):198–200.
•• Shouman K, Vanichkachorn G, Cheshire WP, et al. Autonomic dysfunction following COVID-19 infection: an early experience. Clinical autonomic research: official journal of the Clinical Autonomic Research Society. 2021;31(3):385–94. This case series demonstrated that abnormalities on autonomic testing were seen in the majority of patients with persistent symptoms following COVID-19 infection but were mild in most cases. The most common finding was orthostatic intolerance, often without objective hemodynamic abnormalities on testing.
Stute NL, Stickford JL, Province VM, Augenreich MA, Ratchford SM, Stickford ASL. COVID-19 is getting on our nerves: sympathetic neural activity and haemodynamics in young adults recovering from SARS-CoV-2. J Physiol. 2021;599(18):4269–85.
Banerjee D, Roeker LE, Grogan M, et al. Outcomes of patients with familial transthyretin amyloidosis after liver transplantation. Prog Transplant. 2017;27(3):246–50.
Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–34.
Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–14.
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92.
Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–24.
Xia X, Wang R, Vetrano DL, et al. From normal cognition to cognitive impairment and dementia: impact of orthostatic hypotension. Hypertension. 2021;78(3):769–78.
•• Fedorowski A, Ricci F, Hamrefors V, et al. Orthostatic hypotension: management of a complex, but common, medical problem. Circ Arrhythm Electrophysiol. 2022;15(3): e010573. This is an excellent review focusing on a pathophysiological classification of orthostatic hypotension.
Park JW, Okamoto LE, Shibao CA, Biaggioni I. Pharmacologic treatment of orthostatic hypotension. Autonomic neuroscience : basic & clinical. 2020;229: 102721.
Palma JA, Norcliffe-Kaufmann L, Martinez J, Kaufmann H. Supine plasma NE predicts the pressor response to droxidopa in neurogenic orthostatic hypotension. Neurology. 2018;91(16):e1539–44.
Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol. 2006;63(4):513–8.
Shibao C, Raj SR, Gamboa A, et al. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension. 2007;50(1):47–53.
Shibao CA, Palma JA, Celedonio JE, Martinez J, Kaufmann H, Biaggioni I. Predictors of the pressor response to the norepinephrine transporter inhibitor, atomoxetine, in neurogenic orthostatic hypotension. Hypertension. 2021;78(2):525–31.
Ramirez CE, Okamoto LE, Arnold AC, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2014;64(6):1235–40.
• Kaufmann H, Vickery R, Wang W, et al. Safety and efficacy of ampreloxetine in symptomatic neurogenic orthostatic hypotension: a phase 2 trial. Clin Auton Res: Off J Clin Auton Res Soc. 2021;31(6):699–711. This study demonstrated that ampreloxetine was well tolerated and improved orthostatic symptoms and seated/standing blood pressure with little change in supine blood pressure. Further studies are necessary to investigate if ampreloxetine is beneficial for the treatment of neurogenic orthostatic hypotension in specific subgroups of patients.
Theravance Biopharma, Inc. announces top-line results from a phase 3 study of ampreloxetine in patients with symptomatic neurogenic orthostatic hypotension [Press release]. 2021. https://wwwprnewswirecom/news-releases/theravance-biopharma-inc-announces-top-line-results-from-a-phase-3-study-of-ampreloxetine-in-patients-with-symptomatic-neurogenic-orthostatic-hypotension-301377200html. Accessed 1 Nov 2022.
•• Juraschek SP, Hu JR, Cluett JL, et al. Effects of intensive blood pressure treatment on orthostatic hypotension: a systematic review and individual participant-based meta-analysis. Ann Intern Med. 2021;174(1):58–68. This systematic review and meta-analysis demonstrated that intensive blood pressure-lowering treatment decreases risk for orthostatic hypotension. Orthostatic hypotension, before or in the setting of more intensive blood pressure treatment, should not be viewed as a reason to avoid or de-escalate treatment for hypertension.
Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation. 2007;115(3):387–97.
Fanciulli A, Jordan J, Biaggioni I, et al. Consensus statement on the definition of neurogenic supine hypertension in cardiovascular autonomic failure by the American Autonomic Society (AAS) and the European Federation of Autonomic Societies (EFAS): Endorsed by the European Academy of Neurology (EAN) and the European Society of Hypertension (ESH). Clin Auton Res: Off J Clin Auton Res Soc. 2018;28(4):355–62.
Shannon J, Jordan J, Costa F, Robertson RM, Biaggioni I. The hypertension of autonomic failure and its treatment. Hypertension. 1997;30:1062–7.
•• Newman L, O’Connor JD, Romero-Ortuno R, Reilly RB, Kenny RA. Supine hypertension is associated with an impaired cerebral oxygenation response to orthostasis: finding from the Irish longitudinal study on ageing. Hypertension. 2021;78(1):210–9. This study demonstrated that in a large cohort of community-dwelling older adults, supine hypertension was associated with a larger drop and impaired recovery of cerebral oxygenation upon standing when standardized to orthostatic mean arterial pressure.
Jordan J, Fanciulli A, Tank J, et al. Management of supine hypertension in patients with neurogenic orthostatic hypotension: scientific statement of the American Autonomic Society, European Federation of Autonomic Societies, and the European Society of Hypertension. J Hypertens. 2019;37(8):1541–6.
• Okamoto LE, Celedonio JE, Smith EC, et al. Local passive heat for the treatment of hypertension in autonomic failure. J Am Heart Assoc. 2021;10(7): e018979. This study demonstrated that local heat therapy effectively lowered overnight blood pressure in patients with autonomic failure and supine hypertension and offers a novel approach to treat this condition.
Biaggioni I, Shibao CA, Diedrich A, Muldowney JAS 3rd, Laffer CL, Jordan J. Blood pressure management in afferent baroreflex failure: JACC review topic of the week. J Am Coll Cardiol. 2019;74(23):2939–47.
Norcliffe-Kaufmann L, Palma JA, Martinez J, Kaufmann H. Carbidopa for afferent baroreflex failure in familial dysautonomia: a double-blind randomized crossover clinical trial. Hypertension. 2020;76(3):724–31.
Vinik AI, Maser RE, Ziegler D. Autonomic imbalance: prophet of doom or scope for hope? Diabet Med. 2011;28(6):643–51.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Guillaume Lamotte serves as the managing editor of the journal Clinical Autonomic Research. Paola Sandroni declares no competing interests.
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Autonomic Dysfunction
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lamotte, G., Sandroni, P. Updates on the Diagnosis and Treatment of Peripheral Autonomic Neuropathies. Curr Neurol Neurosci Rep 22, 823–837 (2022). https://doi.org/10.1007/s11910-022-01240-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-022-01240-4