
Abstract
Purpose of Review
The renin-angiotensin system (RAS) plays an important role in modulating cardiovascular function and fluid homeostasis. While the systemic actions of the RAS are widely accepted, the role of the RAS in the brain, its regulation of cardiovascular function, and sympathetic outflow remain controversial. In this report, we discuss the current understanding of central RAS on blood pressure (BP) regulation, in light of recent literature and new experimental techniques.
Recent Findings
Studies using neuronal or glial-specifc mouse models have allowed for greater understanding into the site-specific expression and role centrally expressed RAS proteins have on BP regulation. While all components of the RAS have been identified in cardiovascular regulatory regions of the brain, their actions may be site specific. In a number of animal models of hypertension, reduction in Ang II-mediated signaling, or upregulation of the central ACE2/Ang 1–7 pathway, has been shown to reduce BP, via a reduction in sympathetic signaling and increase parasympathetic tone, respectively. Emerging evidence also suggests that, in part, the female protective phenotype against hypertension may be due to inceased ACE2 activity within cardiovascular regulatory regions of the brain, potentially mediated by estrogen.
Summary
Increasing evidence suggests the importance of a central renin-angiotensin pathway, although its localization and the mechanisms involved in its expression and regulation still need to be clarified and more precisely defined.
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Similar content being viewed by others


Introduction
Hypertension (HTN) is one of the leading causes of morbidity and mortality globally [1]. It accounts for 9.4 million deaths worldwide annually, with over half of all strokes and cases of ischemic heart disease attributable to high blood pressure (BP) [1, 2].
For the majority of patients, the development of HTN is dependent on a combination of genetic and environment factors. In many clinical and animal models, HTN presents with increased sympathetic outflow [3]. The increased sympathetic drive to the cardiovascular system and resultant increase in BP are termed “neurogenic hypertension.” Disturbances in the central nervous system control of sympathetic outflow may induce neurogenic HTN. However, the pathophysiological mechanisms responsible for the increased sympathetic drive in HTN are unknown. The pathogenesis of HTN is complex and undoubtedly caused by multiple factors that regulate heart rate, cardiac output, and total peripheral resistance. Even after over 100 years of study, the mechanisms underlying the development of HTN are incompletely understood.
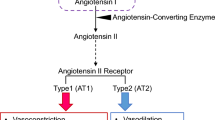
The renin-angiotensin system (RAS) plays an important role in cardiovascular homeostasis, body fluid regulation, and electrolyte balance and has recently been implicated as a metabolic regulator. The RAS has been shown to be key in the development of HTN [4,5,6,7] The classical RAS pathway, in brief, is composed of angiotensinogen (AGT), a precursor, synthetized in the liver, and renin, an aspartyl protease, released by the juxtaglomerular cells of the kidney (Fig. 1). Renin cleaves AGT generating the inactive decapeptide angiotensin I (Ang I). Ang I is activated by the angiotensin converting enzyme (ACE), located on endothelial cells of the lung and kidneys. ACE cleaves the carboxy-terminal His-Leu dipeptide of Ang I to generate the active octapeptide Ang II, the predominant effector protein of the RAS pathway. Binding of Ang II to the angiotensin II type 1 (AT1) receptor results in an increase in BP by inducing vasoconstriction, increases renal sodium reabsorption, and induces the release of aldosterone and arginine vasopressin (AVP) from the adrenal and pituitary glands, respectively, in addition to increasing central sympathetic outflow [8,9,10]. Two subtypes of AT1 receptors (AT1a and AT1b) have been identified in rodents [11, 12]; however, their distinct physiological actions have yet to be clarified. Conversely, Ang II binding to AT2 receptors induces vasodilation, apoptosis, cellular proliferation, and sympatho-inhibition, reduces sodium reabsorptium, and inhibits AVP release [8, 13,14,15,16,17,18,19,20,21,22,23,24,25,26].

The classical and systemic renin angiotensin system. ATG: angiotensinogen, Ang I: angiotensin I, Ang II: angiotensin II, Ang III: angiotensin III, Ang IV: Angiotensin IV, ACE: angiotensin converting enzyme, AMN A: aminopeptidase A, AMN N: aminopeptidase N, AT1R: AT1 receptor, AT2R: AT2 receptor, AT4R: AT4 receptor, DAP: dipepdyl aminopeptidase, Ren: Renin
Ang II is metabolized by aminopeptidase (AMN) A into Ang III (Ang 2–8), which is converted by AMN N into Ang IV (Ang 3–8). Alternatively, Ang II can be metabolized by ACE 2, generating Ang 1–7. Similar to Ang II, Ang III and its metabolites play an important role in AVP and aldosterone release, sympathetic hyperactivity, and BP regulation [19, 27,28,29,30]. However, Ang III has been shown to reduce sodium reabsorbance [31] and has a cardioprotective effect via AT2R [32]. Ang IV affects cognitive function, reduces neuronal apoptosis, and pro-inflammatory [33,34,35,36,37]. Ang 1–7 binds mainly to the Mas receptor whose actions counterbalance many of the deleterious effects of Ang II [38]. Therefore, ACE 2 and Ang 1–7 have been generally thought to be protective to the cardiovascular system.
In addition to endocrine and systemic actions of the RAS, which are now well described, all components of the RAS have now been identified in the brain. Although previously controversial, a number of recent articles have extensively reviewed the current literature on RAS control of central BP regulation providing positive evidence for its importance [23, 39,40,41,42,43,44,45]. A number of animal models have demonstrated the importance of the brain RAS on development and maintenance of HTN [46,47,48,49,50,51,52]. In addition, peripheral Ang II may gain entry into the central nervous system at sites with an intact blood-brain barrier [53]. Recent novel genetic and imaging techniques have allowed a greater understanding of the central RAS activity. This review will provide a broad overview of some of the most recent findings in this field.
Brain RAS/Systemic RAS Interactions
The difficulties in differentiating circulatory versus tissue RAS products limit the comprehension of both systems. However, genetic tools to generate transgenic animals targeting RAS tissue-specific products provide an important tool to dissect these systems.
In a recent review by Nakagawa and Sigmund [39], the authors point out that systemic BP regulation and renal-fluid regulation have generally been attributed to the circulating RAS [54]. Intrarenal Ang II is elevated in Ang II-induced HTN, independent of renal renin levels. The augmentation of intrarenal Ang II is due, in part, to uptake of circulating Ang II via an AT1R mechanism and to endogenous production of Ang II [55]. However, central Ang II activation induces sympathetic outflow (neurotransmission) [56,57,58,59]. AVP release has been attributed to tissue-specific RAS [23, 58, 60, 61]. The existence of both a local centrally acting and a peripheral systemically acting RAS makes understanding the intereactions and regulation between these two systems complex.
Neuronal and Glial RAS
Angiotensinogen
Astrocytes are the main site of AGT synthesis in the brain [62,63,64,65,66] and have been shown to contribute to elevated BP in rodents [50, 51] (Fig. 2). Moreover, AGT expression is responsive to Ang II in both SHR and Wistar astrocytes in vitro [67]. Its absence induces diabetes insipidus [68], and low glial AGT expression has been shown to play a role in the maintenance of diastolic function and exercise tolerance in rats [69].

Brain renin angiotensin system. An overview of the RAS components in central neurons, astrocytes, and microglial cells. ATG: angiotensinogen, Ang I: angiotensin I, Ang II: angiotensin II, Ang III: angiotensin III, Ang IV: Angiotensin IV, ACE: angiotensin converting enzyme, ACE2: angiotensin converting enzyme 2, AT1R: AT1 receptor, AT2R: AT2 receptor, MASR: Mas receptor, PRR: pro-renin receptor
There is also evidence for AGT expression in neurons [50, 65, 70,71,72]. Recently, Agassandian et al. [73] identified a population of neurons in the subfornical organ (SFO) that release AGT and possibly Ang I/II into the cerebral ventricle, when AGT and renin were overexpressed in two different transgenic mouse models.
To demonstrate the differential contribution of neuronal or glial cell AGT in sympathoexcitation, Sakai et al. [71] used a transgenic mouse model that expressed both renin and AGT genes under either neuronal- or glial-specific promoters. The authors showed that Ang II expression in the glial-specific renin/AGT overexpression mice was predominantly observed in the PVN and NTS, whereas in the neuron-specific mice, expression was predominatly observed in the RVLM. They showed that neuronal- and glial-specific Ang II generation exhibited different modulatory effects on arterial baroreflex function, a marker of sympathetic function, possibly due to regional differences in Ang II production.
Renin and Prorenin Receptor
Renin is the predominant rate-limiting step of the RAS cascade and was first identified in the brain in 1971 [74, 75]. However, in 1999, Lee-Kirsch et al. described a new isoform of renin, Renin-b, which is exclusively expressed in the brain. Renin-b evolves from an alternative promoter within exon 1 of the renin gene resulting in the loss of a signal peptide that allows the peptide to be constitutively active and remains intracellular [76]. The existence of intracellular renin is compelling to support the hypothesis that Ang II acts as a neurotransmitter [77].
Renin-b is expressed mainly in neurons [66, 78, 79] and has been identified in cardiovascular regulatory areas including the SFO, paraventricular nucleus (PVN), area postrema (AP), and rostral ventrolateral medulla (RVLM) [80, 81]. Centrally, its expression levels are significantly lower than in the periphery, making it difficult to study in the brain. However, the advancement of genetic tools in recent years has allowed for greater clarity in renin-b localization and function.
The importance of renin-b in the central RAS was indirectly demonstrated by Xu et al. (2011) [82]. These investigators knocked out secreted neuronal- and glial-renin, which had no effect on BP, HR, food, water and sodium intake, renal function, or metabolic rate. These data demonstrate that the secreted version of renin (Renin-a) is dispensable within the brain for cardiovascular, fluid, and metabolic homeostasis. Knockout of renin-b resulted in an increase in BP and sympathetic nerve activity and impaired baroreflex sensitivity as demonstrated by Shinohara et al. [6]. These authors proposed the hypothesis that in the brain, renin-b inhibits renin-a expression under normal conditions. However, in adverse conditions when Renin-b is reduced, renin-a is upregulated and initiates the RAS cascade to induce HTN. This idea is further supported by the observation that in response to deoxycorticosterone acetate (DOCA)–salt treatment to induce HTN, a switch in the expression of renin-b to renin-a has been shown [83].
In a recent controversial study, van Thiel et al. [41] failed to detect any level of renin mRNA (secreted or intracellular) from brain tissue of wild type, DOCA-salt treated, or Ang II infused mice. Moreover, the authors showed an increase in Ang II activity in renin knockout mice, suggesting that Ang II formation might occur by other proteases, such as cathepsin [84, 85]. In addition to the above cardiovascular effects, Shinohara et al. [6] recently demonstrated that Renin-b is also important in the regulation of energy homeostasis and thermogenesis. The authors showed an increase in resting metabolic rate and SNA to brown adipose tissue in renin-b null mice.
Renin-b can bind to a single transmembrane domain of the pro-renin receptor (PRR) [86] resulting in AGT-Ang II cleaving and RAS activation. The PRR is highly expressed in neurons, microgial, and astrocytes in the PVN, SFO, NTS, and area postrema [35, 86, 87]. PRR receptor overexpression is observed in hypertensive animal models [46, 87] and is thought to be regulated either directly by Ang II via an AT1 receptor-dependent pathway [86, 88] or indirectly via COX-2/PGE2 [89].
The expression of the PRR in glial cells appears to be related to pro-inflammatory cytokine release and has been shown to contribute to neuroinflammation and the development of neurogenic HTN [90]. Moreover, neuron-specific PRR deletion reduced DOCA salt-induced HTN in knockout mice by reducing sympathetic tone and was associated with a reduction in Ang II levels [88]. These studies suggest that the PRR plays an important role in the central regulation of BP, although the presence of renin in the brain and the mechanisms involved in its expression still need to be clarified.
Angiotensin Receptors
Angiotensin receptors are expressed in multiple brain regions and have been largely reviewed [39, 40, 91]. Recent data showing optogenetic stimulation of AT1a expressing cells in the parvocellular neurosecretory neurons of the PVN promoted a rise in systolic BP, as well as activation of the hypothalamic-pituitary-adrenal and hypothalamic-pituitary-thyroid axes [23]. In addition, the presence of the AT2R in neurons at the prefrontal cortex, MnPO, portions of the amygdala, NTS and the AP, suggests a modulation not just of cardiovascular function but also of metabolism and stress responses [23].
Angiotensin-Converting Enzyme
Centrally, ACE is predominantly localized to within cerebral vasculature endothelium. However, lower expression levels have also been identified in neurons and astrocytes of cardiovascular regulatory regions including the choroid plexus, SFO, OVLT, hypothalamus, and basal ganglia [92,94,94].
Recently, Faulk et al. showed that ACE is increased in the median preoptic nucleus (MnPO) after 1 week of chronic intermittent hypoxia [95]. The MnPO projects to the PVN and modulates BP regulation. In this study, ACE was not localized to astrocytes.
Injection of human ACE into the brain of Sprgue-Dawley rats increased BP, sympathetic nerve activity, and Ang II levels in cerebral spinal fluid [96]. It has been suggested that increases in central ACE activity or expression occur in hypertension, although the mechanisms behind this are unknown [97, 98].
ACE inhibition increases ACE mRNA expression both within the whole brain and isolated neuronal cultures, suggesting that neuronal ACE can contribute to the central ACE overexpression seen with ACE inhibition [99]. Although the exact role of cell type specific ACE expression within the brain and their role in BP regulation is still unknown, more recently, focus has shifted to examing the role of ACE2 in central BP regulation.
Angiotensin Converting Enzyme 2 and Ang 1–7 in BP Regulation
ACE2 transforms Ang II into Ang 1–7 by cleaving the carboxyterminal phenylalanine residue. Ang 1–7 signals predominantly through Mas receptors, but can also signal through the AT2R. In the peripheral circulation, Ang 1–7 is thought to predominantly regulate BP via nitric oxide (NO)-induced vasodilator effects [100, 101]. Centrally, the role of ACE2 and Ang 1–7 on BP regulation is less well understood. Central ACE2 has been shown to modulate NO synthesis, spontaneous baroreflex sensitivity (sBRS), and parasympathetic tone, suggesting a role for the development of neurogenic hypertension [14, 18, 19, 102, 103]. ACE2 expression and activity have been identified in multiple cardiovascular areas in the brain [47] and are thought to be the predominant enzyme involved in hydrolyzing central Ang II to Ang 1–7 in humans [104]. Central ACE2 downregulation has been observed in a number of animal hypertensive models [102, 105, 106]. On the other hand, neuronal overexpression of ACE2 has been shown to attenuate high BP in a multiple hypertensive models, including Ang II-induced, DOCA salt, and in the SHR [105, 107, 108].
Overexpression of ACE2 in the brain using a mouse model that expresses human ACE2 under a synapsin promoter, syn-hACE2 (SA), demonstrated that ACE2 overexpression attenuated the development of neurogenic hypertension, in a Ang II infusion model. This was partially mediated by preventing the decrease in sBRS and in parasympathetic tone usually mediated by Ang II in this model. Interstingly, no effect on sympathetic outflow was observed [103]. This protective effect of syn-hACE2 upregulation is proposed to be partially mediated through upregulation in expression and phosphorylation of nitric oxide synthase (NOS) in key BP regulation centers of the brain, such as the NTS and RVLM. This is consistent with early reports that showed Ang 1–7 increases NO release in the brain [109] and reinforces baroreflex sensitivity [110].
The role of ACE2/Ang 1–7 in central RAS and BP regulation has remained controversial. While predominantly thought of as producing depressor and bradycardic effects [111], Ang 1–7 appears to have a site-specific action within the brain. When injected into the PVN, Ang 1–7 enhances sympathetic outflow and cardiac sympathetic reflexes to a level comparable to that of Ang II infusion [112]. Previous methods of measuring ACE2 activity by determining Ang 1–7 formation from radio labeled Ang II [113, 114], or traditional fluorescent substrate techniques were nonspecific and difficult to quantify [115, 116]. New methods utilize a quenched fluorescent substrate of ACE2. ACE2 hydrolysis of a proline-lysine peptide bond removes the quenching effect, in the presence or absence of the ACE2 inhibitor DX600. This allows for an easy to use measurement of ACE2 activity across a variety of tissue and cell types [115], although DX600 may have issues with ACE2 sensitivity across different species [117].
This technique has recently been used by Xu et al. to determine if the downregulation of central ACE2 observed in HTN is due to increased ACE2 shedding into the cerebrospinal fluid (CSF) [106, 118, 119]. ACE2 is a transmembrane protein with the N-terminus located extracellularly and the C-terminus intracellularly [120]. The cleavage of the extracellular domain of ACE2 could result in the shed form of ACE2 (sACE2) being detectable in CSF. It is unknown if sACE2 has the same enzymatic activity as the membrane anchored form but it is proposed not to [119]. In this study, increased sACE2 was observed in the CSF of hypertensive individuals and positively correlated with systolic BP. This was normalized in individuals on antihypertensive treatments, which resulted in controlled HTN. The authors established that reduced ACE2 activity, measured using fluorescent quenching in the hypothalamus of the DOCA salt mouse, was correlated with an increase in ADAM17 (ADAM metallopeptidase domain 17) activity. ADAM17 is a disintegrin and metalloprotease originally described as a key sheddase in cytokine formation but is known to cleave a variety of membrane-bound proteins [121]. Its activity levels are dependent on the AT1R, not the elevation of BP [119]. Neither elevating BP with norepinephrine infusion nor in a central AT1 receptor knockout in the DOCA mouse model resulted in increased central ADAM17 activity compared to control [119]. Although the correlation between loss of central ACE2, increased sACE2, and increased ADAM17 within this study appears strong, there was no direct link showing that ADAM17 cleaved ACE2. Elevated sACE2 in human heart failure [122, 123], HTN [123], and severe acute respiratory syndrome [124] have been reported. It is possible that the loss of this compensatory arm of the central RAS axis may be contributing to sympatho-excitation, increased BP, and a negative prognostic outcome in these conditions. Preservation of ACE2 enzyme activity may be critical to inhibition of the Ang II-AT1R axis in the CNS.
Interestingly, in a corollary of the hypertensive state, the involvement of central ACE2 has been demonstrated in rabbits with pacing-induced heart failure in the RVLM and PVN [125] where its protein and message were reduced compared to sham control animals. A follow-up study from the same investigators [126] showed that chronic intracerebroventricular infusion of Ang 1–7 resulted in a reduction in sympathetic nerve activity and enhanced arterial baroreflex function. Xiao et al. [127] clearly showed that heart failure mice that overexpress the human isoform of ACE 2 in the brain exhibit reduced sympathetic outflow as measured by NE excretion and direct recording of RSNA.
These data suggest that in cardiovascular states characterized by increased sympathetic outflow, ACE2 and Ang 1–7 play an important modulatory role. While the cellular mechanisms of the sympatho-inhbition are not completely clear, it is likely that a reduction in oxidative stress plays a role [128].
Sex Differences in Central RAS Regulation
Sex differences in BP regulation and in the progression of HTN have been shown in both clinical and animal models [129,131,131]. Female sex hormones, especially estrogen, have been shown to have a protective effect against the development of HTN and heart failure by direct modulation of the RAS, in the kidney, heart, vasculature, and CNS [130]. Recent studies have observed that brain expression of Ang 1–7, Mas receptors, and neuronal NOS is controlled by female sex hormones, and this may explain the reduced progression from HTN to other CV diseases in females [132].
Historically, studies on animal models of HTN have been carried out in males due to variances in hormonal cycles in females. This strategy has led to conflicting and inconsistent results in mechanisms related to neurogenic HTN in females. The National Institutes of Health and other major funding bodies have emphasized the need to carry out studies in both sexes in order to better define mechanistic differences between sexes [133]. Both classical estrogen receptor subtypes (ERα and ERβ) are expressed in key BP regulatory regions of the brain [134]. Estrogen replacement therapy is cardio protective in postmenopausal women and has previously been shown to decrease Ang II AT1R receptor binding and mRNA within the hypothalamus of female rats [135]. Several studies have examined the effects of sex differences on central Ang II-mediated neurogenic hypertension [130, 136, 137]. Moreover, a number of previously identified pre-conditioning stressors that sensitize rodents to subsequent Ang II-mediated hypertension [39] are thought now to be sex specific. When male rats are pre-treated with a sub-pressor dose of Ang II, subcutaneous or intracerebroventriular, prior to implantation of an osmotic minipump that will infuse the full dose of Ang II used to elicit Ang II-mediated hypertension, they developed a greater rise in BP than those rats that did not receive the sub-pressor Ang II pretreatment [138]. In this study, pretreatment with a sub-pressor dose Ang II resulted in increased expression of renin, Ang II, AT1R, AT2R, and ACE in the lamina terminalis of the forebrain. Conversely, females showed a slight reduction in BP during the pre-treatment with subpressor Ang II, and a significant but lower rise in BP during the high dose Ang II infusion phase which matched the BP rise seen in animals that had only received saline in the induction phase [136]. Interestingly, ovariectomized (OVX) females responded similar to the males, showing no change in BP during the induction phase, but a significant increase in BP during pressor phase. It was hypothesized that this difference in response between males and females was due to increased Ang 1–7-mediated signaling in the central nervous system (CNS) of females. Intracerebroventricular infusion of the Ang 1–7 antagonist A-779 in female rats during the induction phase prevented the attenuation in BP increase [136]. This suggests that part of the protective role of estrogen may be mediated by the central effects of Ang 1–7. Intracerebroventricular infusion of Ang 1–7 in male and OVX female rats reduced the induction of HTN but did not completely abolish it. If increased Ang 1–7 signaling mediates reduced HTN in females, it would be of interest to see if females have increased central ACE2 levels and a reduction in ACE2 shedding compared to male hypertensive animals.
Estrogen has previously been shown to prevent downregulation of expression of ACE and AT1R mRNA in the kidney, lung, aorta, heart, adrenal, and the PVN [139, 140]. The studies mentioned above imply estrogen modulate the Ang 1–7 pathway, and therefore would be implicated in preventing Ang II-mediated reduction in vagal tone [141]. Previous studies have suggested the antihypertensive effects of estrogen in the CNS to be due to attenuation of sympathetic outflow, although this appears to be region specific [142, 143]. Estrogen injection into the NTS or RVLM of male rats decreased BP and augmented baroreflex control of sympathetic nerve activity, whereas injection into the nucleus ambiguus, augmented baroreflex control of HR and increased vagal nerve activity, resulting in reduced HR with no change in BP [144]. Estrogen attenuation of sympathetic outflow is at least partially due to increase NO signaling. nNOS expression is higher in the SFO and PVN of female rats compared to male, and Ang II infusion further increased nNOS expression in intact females, with no change in expression in OVX females or males [145]. Estrogen has also been implicated in reducing sympathetic outflow by reducing central ROS production. Estrogen injected into the SFO inhibited Ang II induced hypertension, SFO nerve signaling, and intracellular ROS production [146]. Where increasing evidence suggests the importance of estrogen in modulating sex differences observed in neurogenic hypertension, its exact role and regional specificity still remain to be seen.
Summary
The RAS is highly complex, modulating cardiovascular, neuroendocrine, sodium homeostasis, and metabolism. Thought once to predominantly act systemically through a number of endocrine, paracrine, and intracrine effects, the bulk of the evidence points to the role of a centrally acting RAS pathway which participates in the sympathetic hyperactivity observed in HTN and other cardiovascular disease states. The true role of the central RAS pathway and its regulation in both physiological and pathophysiological states is an active area of investigation. Recent evidence suggests a constitutively active brain-specific isoform of renin, renin-b that may regulate the central RAS axis. All components of the RAS have been located in central cell types including neurons, glia, and cerebral blood vessels. The stimuli (neuronal or humoral) that regulate each component are still unclear. Figure 2 shows the central components of the RAS and the potenitial cell locations of each component based on our current knowledge. Finally, some of the sex differences observed in neurogenic hypertension may be due to differential regulation of Ang 1–7 in females.
References
Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart disease and stroke Statistics-2016 update: A report from the American Heart Association. Circulation. 2016;133(4):e38–360. https://doi.org/10.1161/CIR.0000000000000350.
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: A systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2224–60. https://doi.org/10.1016/S0140-6736(12)61766-8.
Grassi G, Ram VS. Evidence for a critical role of the sympathetic nervous system in hypertension. J Am Soc Hyper : JASH. 2016;10(5):457–66. https://doi.org/10.1016/j.jash.2016.02.015.
Kouyama R, Suganami T, Nishida J, Tanaka M, Toyoda T, Kiso M, et al. Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a receptor. Endocrinology. 2005;146(8):3481–9. https://doi.org/10.1210/en.2005-0003.
Claflin KE, Sandgren JA, Lambertz AM, Weidemann BJ, Littlejohn NK, Burnett CM, et al. Angiotensin AT1A receptors on leptin receptor-expressing cells control resting metabolism. J Clin Invest. 2017;127(4):1414–24. https://doi.org/10.1172/JCI88641.
Shinohara K, Nakagawa P, Gomez J, Morgan DA, Littlejohn NK, Folchert MD, et al. Selective Deletion of Renin-b in the Brain Alters Drinking and Metabolism. Hypertension (Dallas, Tex : 1979). 2017;70(5):990–7. https://doi.org/10.1161/HYPERTENSIONAHA.117.09923.
Littlejohn NK, Keen HL, Weidemann BJ, Claflin KE, Tobin KV, Markan KR, et al. Suppression of resting metabolism by the angiotensin AT2 receptor. Cell Rep. 2016;16(6):1548–60. https://doi.org/10.1016/j.celrep.2016.07.003.
Gao J, Marc Y, Iturrioz X, Leroux V, Balavoine F, Llorens-Cortes C. A new strategy for treating hypertension by blocking the activity of the brain renin-angiotensin system with aminopeptidase a inhibitors. Clin Sci (Lond). 2014;127(3):135–48. https://doi.org/10.1042/CS20130396.
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4(3):1201–28. https://doi.org/10.1002/cphy.c130040.
Yang R, Smolders I, Dupont AG. Blood pressure and renal hemodynamic effects of angiotensin fragments. Hypertens Res. 2011;34(6):674–83. https://doi.org/10.1038/hr.2011.24.
Iwai N, Inagami T. Identification of two subtypes in the rat type I angiotensin II receptor. FEBS Lett. 1992;298(2–3):257–60.
Kakar SS, Riel KK, Neill JD. Differential expression of angiotensin II receptor subtype mRNAs (AT-1A and AT-1B) in the brain. Biochem Biophys Res Commun. 1992;185(2):688–92.
Yayama K, Okamoto H. Angiotensin II-induced vasodilation via type 2 receptor: Role of bradykinin and nitric oxide. Int Immunopharmacol. 2008;8(2):312–8. https://doi.org/10.1016/j.intimp.2007.06.012.
Savoia C, Ebrahimian T, He Y, Gratton JP, Schiffrin EL, Touyz RM. Angiotensin II/AT2 receptor-induced vasodilation in stroke-prone spontaneously hypertensive rats involves nitric oxide and cGMP-dependent protein kinase. J Hypertens. 2006;24(12):2417–22. https://doi.org/10.1097/01.hjh.0000251902.85675.7e.
Ou Z, Jiang T, Gao Q, Tian YY, Zhou JS, Wu L et al. Mitochondrial-dependent mechanisms are involved in angiotensin II-induced apoptosis in dopaminergic neurons. J Renin Angiotensin Aldosterone Syst. 2016;17(4). doi:https://doi.org/10.1177/1470320316672349.
Ognibene DT, Oliveira PR, Marins de Carvalho LC, Costa CA, Espinoza LA, Criddle DN, et al. angiotensin II-mediated vasodilation is reduced in adult spontaneously hypertensive rats despite enhanced expression of AT2 receptors. Clin Exp Pharmacol Physiol. 2009;36(1):12–9. https://doi.org/10.1111/j.1440-1681.2008.05054.x.
Goel R, Bhat SA, Hanif K, Nath C, Shukla R. Angiotensin II Receptor blockers attenuate lipopolysaccharide-induced memory impairment by modulation of NF-kappaB-mediated BDNF/CREB expression and apoptosis in spontaneously hypertensive rats. Mol Neurobiol 2017. doi:https://doi.org/10.1007/s12035-017-0450-5.
Cosentino F, Savoia C, De Paolis P, Francia P, Russo A, Maffei A, et al. Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists. Am J Hypertens. 2005;18(4 Pt 1):493–9. https://doi.org/10.1016/j.amjhyper.2004.11.007.
Gao J, Zucker IH, Gao L. Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure. Am J Hypertens. 2014;27(10):1248–56. https://doi.org/10.1093/ajh/hpu044.
Gao L, Wang WZ, Wang W, Zucker IH. Imbalance of angiotensin type 1 receptor and angiotensin II type 2 receptor in the rostral ventrolateral medulla: potential mechanism for sympathetic overactivity in heart failure. Hypertension (Dallas, Tex : 1979). 2008;52(4):708–14. https://doi.org/10.1161/HYPERTENSIONAHA.108.116228.
Gao L, Wang W, Wang W, Li H, Sumners C, Zucker IH. Effects of angiotensin type 2 receptor overexpression in the rostral ventrolateral medulla on blood pressure and urine excretion in normal rats. Hypertension (Dallas, Tex : 1979). 2008;51(2):521–7. https://doi.org/10.1161/HYPERTENSIONAHA.107.101717.
Gao J, Zhang H, Le KD, Chao J, Gao L. Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am J Hypertens. 2011;24(6):724–30. https://doi.org/10.1038/ajh.2011.33.
de Kloet AD, Wang L, Ludin JA, Smith JA, Pioquinto DJ, Hiller H, et al. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct. 2016;221(2):891–912. https://doi.org/10.1007/s00429-014-0943-1.
Ji Y, Wang Z, Li Z, Zhang A, Jin Y, Chen H, et al. Angiotensin II enhances proliferation and inflammation through AT1/PKC/NF-kappaB signaling pathway in hepatocellular carcinoma cells. Cell Physiol Biochem. 2016;39(1):13–32. https://doi.org/10.1159/000445602.
Chao J, Gao J, Parbhu KJ, Gao L. Angiotensin type 2 receptors in the intermediolateral cell column of the spinal cord: Negative regulation of sympathetic nerve activity and blood pressure. Int J Cardiol. 2013;168(4):4046–55. https://doi.org/10.1016/j.ijcard.2013.06.051.
Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT(2) receptor activation induces natriuresis and lowers blood pressure. Circ Res. 2014;115(3):388–99. https://doi.org/10.1161/CIRCRESAHA.115.304110.
Del Borgo M, Wang Y, Bosnyak S, Khan M, Walters P, Spizzo I, et al. beta-Pro7Ang III is a novel highly selective angiotensin II type 2 receptor (AT2R) agonist, which acts as a vasodepressor agent via the AT2R in conscious spontaneously hypertensive rats. Clin Sci (Lond). 2015;129(6):505–13. https://doi.org/10.1042/CS20150077.
Huang BS, Ahmad M, White RA, Marc Y, Llorens-Cortes C, Leenen FH. Inhibition of brain angiotensin III attenuates sympathetic hyperactivity and cardiac dysfunction in rats post-myocardial infarction. Cardiovasc Res. 2013;97(3):424–31. https://doi.org/10.1093/cvr/cvs420.
Yatabe J, Yoneda M, Yatabe MS, Watanabe T, Felder RA, Jose PA, et al. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology. 2011;152(4):1582–8. https://doi.org/10.1210/en.2010-1070.
Yang R, Smolders I, De Bundel D, Fouyn R, Halberg M, Demaegdt H, et al. Brain and peripheral angiotensin II type 1 receptors mediate renal vasoconstrictor and blood pressure responses to angiotensin IV in the rat. J Hypertens. 2008;26(5):998–1007. https://doi.org/10.1097/HJH.0b013e3282f5ed58.
Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension (Dallas, Tex : 1979). 2009;53(2):338–43. https://doi.org/10.1161/HYPERTENSIONAHA.108.124198.
Park BM, Gao S, Cha SA, Park BH, Kim SH. Cardioprotective effects of angiotensin III against ischemic injury via the AT2 receptor and KATP channels. Physiol Rep. 2013;1(6):e00151. https://doi.org/10.1002/phy2.151.
Yeatman HR, Albiston AL, Burns P, Chai SY. Forebrain neurone-specific deletion of insulin-regulated aminopeptidase causes age related deficits in memory. Neurobiol Learn Mem. 2016;136:174–82. https://doi.org/10.1016/j.nlm.2016.09.017.
Paris JJ, Eans SO, Mizrachi E, Reilley KJ, Ganno ML, McLaughlin JP. Central administration of angiotensin IV rapidly enhances novel object recognition among mice. Neuropharmacology. 2013;70:247–53. https://doi.org/10.1016/j.neuropharm.2013.01.025.
Hennrikus MT, Gonzalez AA, Prieto MC. The Prorenin Receptor in the Cardiovascular System and Beyond. Am J Physiol Heart Circ Physiol. 2017:ajpheart 00373 2017. doi:https://doi.org/10.1152/ajpheart.00373.2017.
Moeller I, Small DH, Reed G, Harding JW, Mendelsohn FA, Chai SY. Angiotensin IV inhibits neurite outgrowth in cultured embryonic chicken sympathetic neurones. Brain Res. 1996;725(1):61–6.
Kakinuma Y, Hama H, Sugiyama F, Goto K, Murakami K, Fukamizu A. Anti-apoptotic action of angiotensin fragments to neuronal cells from angiotensinogen knock-out mice. Neurosci Lett. 1997;232(3):167–70.
Nunes-Silva A, Rocha GC, Vaz LN, Faria MH, Simoes e Silva AC. Physical exercise and ACE2-Angiotensin-(1–7)-Mas receptor axis of the Renin Angiotensin System. Protein Pept Lett. 2017. doi:https://doi.org/10.2174/0929866524666170728151401.
Nakagawa P, Sigmund CD. How Is the Brain Renin-Angiotensin System Regulated? Hypertension (Dallas, Tex : 1979). 2017;70(1):10–8. https://doi.org/10.1161/hypertensionaha.117.08550.
Marc Y, Llorens-Cortes C. The role of the brain renin-angiotensin system in hypertension: Implications for new treatment. Prog Neurobiol. 2011;95(2):89–103. https://doi.org/10.1016/j.pneurobio.2011.06.006.
van Thiel BS, Goes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, et al. Brain Renin-Angiotensin System: Does It Exist? Hypertension (Dallas, Tex : 1979). 2017;69(6):1136–44. https://doi.org/10.1161/HYPERTENSIONAHA.116.08922.
Huber G, Schuster F, Raasch W. Brain renin-angiotensin system in the pathophysiology of cardiovascular diseases. Pharmacol Res. 2017;125(Pt A):72–90. https://doi.org/10.1016/j.phrs.2017.06.016.
Farag E, Sessler DI, Ebrahim Z, Kurz A, Morgan J, Ahuja S, et al. The renin angiotensin system and the brain: New developments. J Clin Neurosci. 2017;46:1–8. https://doi.org/10.1016/j.jocn.2017.08.055.
Arnold AC, Gallagher PE, Diz DI. Brain renin-angiotensin system in the nexus of hypertension and aging. Hypertens Res. 2013;36(1):5–13. https://doi.org/10.1038/hr.2012.161.
Coble JP, Grobe JL, Johnson AK, Sigmund CD. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: Importance of the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2015;308(4):R238–49. https://doi.org/10.1152/ajpregu.00486.2014.
Li W, Peng H, Cao T, Sato R, McDaniels SJ, Kobori H, et al. Brain-targeted (pro)renin receptor knockdown attenuates angiotensin II-dependent hypertension. Hypertension (Dallas, Tex : 1979). 2012;59(6):1188–94. https://doi.org/10.1161/HYPERTENSIONAHA.111.190108.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R373–81. https://doi.org/10.1152/ajpregu.00292.2006.
Fukamizu A, Sugimura K, Takimoto E, Sugiyama F, Seo MS, Takahashi S, et al. Chimeric renin-angiotensin system demonstrates sustained increase in blood pressure of transgenic mice carrying both human renin and human angiotensinogen genes. J Biol Chem. 1993;268(16):11617–21.
Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol. 2006;291(5):H2166–72. https://doi.org/10.1152/ajpheart.00061.2006.
Morimoto S, Cassell MD, Sigmund CD. Glia- and neuron-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277(36):33235–41. https://doi.org/10.1074/jbc.M204309200.
Sherrod M, Davis DR, Zhou X, Cassell MD, Sigmund CD. Glial-specific ablation of angiotensinogen lowers arterial pressure in renin and angiotensinogen transgenic mice. Am J Physiol Regul Integr Comp Physiol. 2005;289(6):R1763–9. https://doi.org/10.1152/ajpregu.00435.2005.
Sinnayah P, Lazartigues E, Sakai K, Sharma RV, Sigmund CD, Davisson RL. Genetic ablation of angiotensinogen in the subfornical organ of the brain prevents the central angiotensinergic pressor response. Circ Res. 2006;99(10):1125–31. https://doi.org/10.1161/01.RES.0000250259.66683.f5.
Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: Novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J Physiol. 2016;594(6):1591–600. https://doi.org/10.1113/JP271584.
Navar LG, Imig JD, Zou L, Wang CT. Intrarenal production of angiotensin II. Semin Nephrol. 1997;17(5):412–22.
Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension (Dallas, Tex : 1979). 2002;39(2 Pt 2):316–22.
Xue B, Thunhorst RL, Yu Y, Guo F, Beltz TG, Felder RB, et al. Central Renin-Angiotensin System Activation and Inflammation Induced by High-Fat Diet Sensitize Angiotensin II-Elicited Hypertension. Hypertension (Dallas, Tex : 1979). 2016;67(1):163–70. https://doi.org/10.1161/HYPERTENSIONAHA.115.06263.
Costa M, Majewski H. Facilitation of noradrenaline release from sympathetic nerves through activation of ACTH receptors, beta-adrenoceptors and angiotensin II receptors. Br J Pharmacol. 1988;95(3):993–1001.
Khanmoradi M, Nasimi A. Angiotensin II in the paraventricular nucleus stimulates sympathetic outflow to the cardiovascular system and make vasopressin release in rat. Neurosci Lett. 2016;632:98–103. https://doi.org/10.1016/j.neulet.2016.08.040.
Pellegrino PR, Schiller AM, Haack KK, Zucker IH. Central Angiotensin-II Increases Blood Pressure and Sympathetic Outflow via Rho Kinase Activation in Conscious Rabbits. Hypertension (Dallas, Tex : 1979). 2016;68(5):1271–80. https://doi.org/10.1161/hypertensionaha.116.07792.
Stachniak TJ, Trudel E, Bourque CW. Cell-specific retrograde signals mediate antiparallel effects of angiotensin II on osmoreceptor afferents to vasopressin and oxytocin neurons. Cell Rep. 2014;8(2):355–62. https://doi.org/10.1016/j.celrep.2014.06.029.
Steckelings U, Lebrun C, Qadri F, Veltmar A, Unger T. Role of brain angiotensin in cardiovascular regulation. J Cardiovasc Pharmacol. 1992;19(Suppl 6):S72–9.
Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science. 1988;242(4884):1444–6.
Thomas WG, Sernia C. Immunocytochemical localization of angiotensinogen in the rat brain. Neuroscience. 1988;25(1):319–41.
Campbell DJ, Bouhnik J, Menard J, Corvol P. Identity of angiotensinogen precursors of rat brain and liver. Nature. 1984;308(5955):206–8.
Yang G, Gray TS, Sigmund CD, Cassell MD. The angiotensinogen gene is expressed in both astrocytes and neurons in murine central nervous system. Brain Res. 1999;817(1–2):123–31.
Juillerat-Jeanneret L, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, Maerki HP, et al. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer. 2004;90(5):1059–68. https://doi.org/10.1038/sj.bjc.6601646.
Gowrisankar YV, Clark MA. Regulation of angiotensinogen expression by angiotensin II in spontaneously hypertensive rat primary astrocyte cultures. Brain Res. 2016;1643:51–8. https://doi.org/10.1016/j.brainres.2016.04.059.
Schinke M, Baltatu O, Bohm M, Peters J, Rascher W, Bricca G, et al. Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc Natl Acad Sci U S A. 1999;96(7):3975–80.
Groban L, Wang H, Machado FS, Trask AJ, Kritchevsky SB, Ferrario CM, et al. Low glial angiotensinogen improves body habitus, diastolic function, and exercise tolerance in aging male rats. Cardiovasc Endocrinol. 2012;1(3):49–58. https://doi.org/10.1097/XCE.0b013e32835a2159.
Palkovits M, Mezey E, Fodor M, Ganten D, Bahner U, Geiger H, et al. Neurotransmitters and neuropeptides in the baroreceptor reflex arc: Connections between the nucleus of the solitary tract and the ventrolateral medulla oblongata in the rat. Clin Exp Hypertens. 1995;17(1–2):101–13.
Sakai K, Chapleau MW, Morimoto S, Cassell MD, Sigmund CD. Differential modulation of baroreflex control of heart rate by neuron- vs. glia-derived angiotensin II. Physiol Genomics. 2004;20(1):66–72. https://doi.org/10.1152/physiolgenomics.00168.2004.
Garrido-Gil P, Rodriguez-Perez AI, Fernandez-Rodriguez P, Lanciego JL, Labandeira-Garcia JL. Expression of angiotensinogen and receptors for angiotensin and prorenin in the rat and monkey striatal neurons and glial cells. Brain Struct Funct. 2017;222(6):2559–71. https://doi.org/10.1007/s00429-016-1357-z.
Agassandian K, Grobe JL, Liu X, Agassandian M, Thompson AP, Sigmund CD, et al. Evidence for intraventricular secretion of angiotensinogen and angiotensin by the subfornical organ using transgenic mice. Am J Physiol Regul Integr Comp Physiol. 2017;312(6):R973–R81. https://doi.org/10.1152/ajpregu.00511.2016.
Ganten D, Marquez-Julio A, Granger P, Hayduk K, Karsunky KP, Boucher R, et al. Renin in dog brain. Am J Phys. 1971;221(6):1733–7.
Fischer-Ferraro C, Nahmod VE, Goldstein DJ, Finkielman S. Angiotensin and renin in rat and dog brain. J Exp Med. 1971;133(2):353–61.
Lee-Kirsch MA, Gaudet F, Cardoso MC, Lindpaintner K. Distinct renin isoforms generated by tissue-specific transcription initiation and alternative splicing. Circ Res. 1999;84(2):240–6.
Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: Fact, hypothesis, or fantasy. Physiology (Bethesda). 2008;23:187–93. https://doi.org/10.1152/physiol.00002.2008.
Lavoie JL, Cassell MD, Gross KW, Sigmund CD. Adjacent expression of renin and angiotensinogen in the rostral ventrolateral medulla using a dual-reporter transgenic model. Hypertension (Dallas, Tex : 1979). 2004;43(5):1116–9. https://doi.org/10.1161/01.HYP.0000125143.73301.94.
Allen AM, O'Callaghan EL, Hazelwood L, Germain S, Castrop H, Schnermann J, et al. Distribution of cells expressing human renin-promoter activity in the brain of a transgenic mouse. Brain Res. 2008;1243:78–85. https://doi.org/10.1016/j.brainres.2008.09.046.
Naruse M, Naruse K, McKenzie JC, Schelling P, Inagami T. Regional distribution of renin and angiotensinogen in the brain of normotensive (WKY) and spontaneously hypertensive (SHR) rats. Brain Res. 1985;333(1):147–50.
Schelling P, Meyer D, Loos HE, Speck G, Phillips MI, Johnson AK, et al. A micromethod for the measurement of renin in brain nuclei: Its application in spontaneously hypertensive rats. Neuropharmacology. 1982;21(5):455–63.
Xu D, Borges GR, Davis DR, Agassandian K, Sequeira Lopez ML, Gomez RA, et al. Neuron- or glial-specific ablation of secreted renin does not affect renal renin, baseline arterial pressure, or metabolism. Physiol Genomics. 2011;43(6):286–94. https://doi.org/10.1152/physiolgenomics.00208.2010.
Grobe JL, Rahmouni K, Liu X, Sigmund CD. Metabolic rate regulation by the renin-angiotensin system: Brain vs. body. Pflugers Arch. 2013;465(1):167–75. https://doi.org/10.1007/s00424-012-1096-9.
Deinum J, Derkx FH, Danser AH, Schalekamp MA. Identification and quantification of renin and prorenin in the bovine eye. Endocrinology. 1990;126(3):1673–82. https://doi.org/10.1210/endo-126-3-1673.
Karamyan VT, Speth RC. Enzymatic pathways of the brain renin-angiotensin system: Unsolved problems and continuing challenges. Regul Pept. 2007;143(1–3):15–27. https://doi.org/10.1016/j.regpep.2007.03.006.
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109(11):1417–27. https://doi.org/10.1172/JCI14276.
Shan Z, Shi P, Cuadra AE, Dong Y, Lamont GJ, Li Q, et al. Involvement of the brain (pro)renin receptor in cardiovascular homeostasis. Circ Res. 2010;107(7):934–8. https://doi.org/10.1161/CIRCRESAHA.110.226977.
Li W, Peng H, Mehaffey EP, Kimball CD, Grobe JL, van Gool JM, et al. Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension. Hypertension (Dallas, Tex : 1979). 2014;63(2):316–23. https://doi.org/10.1161/HYPERTENSIONAHA.113.02041.
Wang F, Lu X, Peng K, Du Y, Zhou SF, Zhang A, et al. Prostaglandin E-prostanoid4 receptor mediates angiotensin II-induced (pro)renin receptor expression in the rat renal medulla. Hypertension (Dallas, Tex : 1979). 2014;64(2):369–77. https://doi.org/10.1161/HYPERTENSIONAHA.114.03654.
Shi P, Grobe JL, Desland FA, Zhou G, Shen XZ, Shan Z, et al. Direct pro-inflammatory effects of prorenin on microglia. PLoS One. 2014;9(10):e92937. https://doi.org/10.1371/journal.pone.0092937.
de Kloet AD, Steckelings UM, Sumners C. Protective angiotensin type 2 receptors in the brain and hypertension. Curr Hypertens Rep. 2017;19(6):46. https://doi.org/10.1007/s11906-017-0746-x.
Bodiga VL, Bodiga S. Renin angiotensin system in cognitive function and dementia. Asian J Neurosci. 2013;2013:18. https://doi.org/10.1155/2013/102602.
Whiting P, Nava S, Mozley L, Eastham H, Poat J. Expression of angiotensin converting enzyme mRNA in rat brain. Brain Res Mol Brain Res. 1991;11(1):93–6.
Rogerson FM, Schlawe I, Paxinos G, Chai SY, McKinley MJ, Mendelsohn FA. Localization of angiotensin converting enzyme by in vitro autoradiography in the rabbit brain. J Chem Neuroanat. 1995;8(4):227–43.
Faulk K, Shell B, Nedungadi TP, Cunningham JT. Role of angiotensin-converting enzyme 1 within the median preoptic nucleus following chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2017;312(2):R245–R52. https://doi.org/10.1152/ajpregu.00472.2016.
Nakamura S, Moriguchi A, Morishita R, Yamada K, Nishii T, Tomita N, et al. Activation of the brain angiotensin system by in vivo human angiotensin-converting enzyme gene transfer in rats. Hypertension (Dallas, Tex : 1979). 1999;34(2):302–8.
Zhao X, White R, Huang BS, Van Huysse J, Leenen FH. High salt intake and the brain renin--angiotensin system in dahl salt-sensitive rats. J Hypertens. 2001;19(1):89–98.
Xue B, Yu Y, Zhang Z, Guo F, Beltz TG, Thunhorst RL, et al. Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension (Dallas, Tex : 1979). 2016;67(5):970–6. https://doi.org/10.1161/HYPERTENSIONAHA.115.06736.
King SJ, Oparil S, Berecek KH. Neuronal angiotensin-converting enzyme (ACE) gene expression is increased by converting enzyme inhibitors (CEI). Mol Cell Neurosci. 1991;2(1):13–20.
Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, et al. Angiotensin-converting enzyme 2 and angiotensin 1-7: Novel therapeutic targets. Nat Rev Cardiol. 2014;11(7):413–26. https://doi.org/10.1038/nrcardio.2014.59.
Lin S, Pan H, Wu H, Ren D, Lu J. Role of the ACE2Ang(17)mas axis in blood pressure regulation and its potential as an antihypertensive in functional foods (review). Mol Med Rep. 2017;16(4):4403–12. https://doi.org/10.3892/mmr.2017.7168.
Xia H, Feng Y, Obr TD, Hickman PJ, Angiotensin LE II. type 1 receptor-mediated reduction of angiotensin-converting enzyme 2 activity in the brain impairs baroreflex function in hypertensive mice. Hypertension (Dallas, Tex : 1979). 2009;53(2):210–6. https://doi.org/10.1161/HYPERTENSIONAHA.108.123844.
Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RA, et al. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106(2):373–82. https://doi.org/10.1161/circresaha.109.208645.
Xu C, Lu A, Lu X, Zhang L, Fang H, Zhou L, et al. Activation of Renal (Pro)Renin Receptor Contributes to High Fructose-Induced Salt Sensitivity. Hypertension (Dallas, Tex : 1979). 2017;69(2):339–48. https://doi.org/10.1161/HYPERTENSIONAHA.116.08240.
Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension (Dallas, Tex : 1979). 2007;49(4):926–31. https://doi.org/10.1161/01.hyp.0000259942.38108.20.
Deshotels MR, Xia H, Lazartigues E, Filipeanu CM. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension (Dallas, Tex : 1979). 2014;64(6):1368–75. https://doi.org/10.1161/hypertensionaha.114.03743.
Feng Y, Yue X, Xia H, Bindom SM, Hickman PJ, Filipeanu CM, et al. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ Res. 2008;102(6):729–36. https://doi.org/10.1161/circresaha.107.169110.
Xia H, de Queiroz TM, Sriramula S, Feng Y, Johnson T, Mungrue IN, et al. Brain ACE2 overexpression reduces DOCA-salt hypertension independently of endoplasmic reticulum stress. Am J Physiol Regul Integr Comp Physiol. 2015;308(5):R370–8. https://doi.org/10.1152/ajpregu.00366.2014.
Calka J, Block CH. Angiotensin-(1-7) and nitric oxide synthase in the hypothalamo-neurohypophysial system. Brain Res Bull. 1993;30(5–6):677–85.
Campagnole-Santos MJ, Heringer SB, Batista EN, Khosla MC, Santos RA. Differential baroreceptor reflex modulation by centrally infused angiotensin peptides. Am J Phys. 1992;263(1 Pt 2):R89–94.
Campagnole-Santos MJ, Diz DI, Santos RA, Khosla MC, Brosnihan KB, Ferrario CM. Cardiovascular effects of angiotensin-(1-7) injected into the dorsal medulla of rats. Am J Phys. 1989;257(1 Pt 2):H324–9.
Sun HJ, Li P, Chen WW, Xiong XQ, Han Y. Angiotensin II and angiotensin-(1-7) in paraventricular nucleus modulate cardiac sympathetic afferent reflex in renovascular hypertensive rats. PLoS One. 2012;7(12):e52557. https://doi.org/10.1371/journal.pone.0052557.
Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, et al. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond). 2014;126(7):461–9. https://doi.org/10.1042/CS20130400.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605–10. https://doi.org/10.1161/circulationaha.104.510461.
Sriramula S, Pedersen KB, Xia H, Lazartigues E. Determining the enzymatic activity of angiotensin-converting enzyme 2 (ACE2) in brain tissue and cerebrospinal fluid using a quenched fluorescent substrate. Methods Mol Biol. 2017;1527:117–26. https://doi.org/10.1007/978-1-4939-6625-7_9.
Yan ZH, Ren KJ, Wang Y, Chen S, Brock TA, Rege AA. Development of intramolecularly quenched fluorescent peptides as substrates of angiotensin-converting enzyme 2. Anal Biochem. 2003;312(2):141–7.
Pedersen KB, Sriramula S, Chhabra KH, Xia H, Lazartigues E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am J Physiol Regul Integr Comp Physiol. 2011;301(5):R1293–9. https://doi.org/10.1152/ajpregu.00339.2011.
Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113(9):1087–96. https://doi.org/10.1161/circresaha.113.301811.
Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, et al. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ Res. 2017;121(1):43–55. https://doi.org/10.1161/CIRCRESAHA.116.310509.
Warner FJ, Lew RA, Smith AI, Lambert DW, Hooper NM, Turner AJ. Angiotensin-converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J Biol Chem. 2005;280(47):39353–62. https://doi.org/10.1074/jbc.M508914200.
Xu J, Mukerjee S, Silva-Alves CR, Carvalho-Galvao A, Cruz JC, Balarini CM, et al. A Disintegrin and metalloprotease 17 in the cardiovascular and central nervous systems. Front Physiol. 2016;7:469. https://doi.org/10.3389/fphys.2016.00469.
Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. 2008;52(9):750–4. https://doi.org/10.1016/j.jacc.2008.02.088.
Uri K, Fagyas M, Manyine Siket I, Kertesz A, Csanadi Z, Sandorfi G, et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS One. 2014;9(4):e87845. https://doi.org/10.1371/journal.pone.0087845.
Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L84–96. https://doi.org/10.1152/ajplung.00071.2009.
Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol (Bethesda, Md : 1985). 2010;108(4):923–32. https://doi.org/10.1152/japplphysiol.00840.2009.
Kar S, Gao L, Belatti DA, Curry PL, Zucker IH. Central angiotensin (1–7) enhances baroreflex gain in conscious rabbits with heart failure. Hypertension (Dallas, Tex : 1979). 2011;58(4):627–34. https://doi.org/10.1161/hypertensionaha.111.177600.
Xiao L, Gao L, Lazartigues E, Zucker IH. Brain-selective overexpression of angiotensin-converting enzyme 2 attenuates sympathetic nerve activity and enhances baroreflex function in chronic heart failure. Hypertension (Dallas, Tex : 1979). 2011;58(6):1057–65. https://doi.org/10.1161/hypertensionaha.111.176636.
Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, et al. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One. 2011;6(7):e22682. https://doi.org/10.1371/journal.pone.0022682.
Sandberg K, Ji H. Sex differences in primary hypertension. Biol Sex Differ. 2012;3(1):7. https://doi.org/10.1186/2042-6410-3-7.
Xue B, Johnson AK, Hay M. Sex differences in angiotensin II- and aldosterone-induced hypertension: The central protective effects of estrogen. Am J Physiol Regul Integr Comp Physiol. 2013;305(5):R459–63. https://doi.org/10.1152/ajpregu.00222.2013.
Hay M, Xue B, Johnson AK. Yes! Sex matters: Sex, the brain and blood pressure. Curr Hypertens Rep. 2014;16(8):458. https://doi.org/10.1007/s11906-014-0458-4.
Cheng Y, Li Q, Zhang Y, Wen Q, Zhao J. Effects of female sex hormones on expression of the Ang-(1-7)/mas-R/nNOS pathways in rat brain. Can J Physiol Pharmacol. 2015;93(11):993–8. https://doi.org/10.1139/cjpp-2015-0087.
Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. 2014;509(7500):282–3.
Spary EJ, Maqbool A, Batten TF. Oestrogen receptors in the central nervous system and evidence for their role in the control of cardiovascular function. J Chem Neuroanat. 2009;38(3):185–96. https://doi.org/10.1016/j.jchemneu.2009.05.008.
Kisley LR, Sakai RR, Fluharty SJ. Estrogen decreases hypothalamic angiotensin II AT1 receptor binding and mRNA in the female rat. Brain Res. 1999;844(1–2):34–42.
Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am J Physiol Heart Circ Physiol. 2014;307(2):H191–8. https://doi.org/10.1152/ajpheart.01012.2013.
Dai SY, Peng W, Zhang YP, Li JD, Shen Y, Sun XF. Brain endogenous angiotensin II receptor type 2 (AT2-R) protects against DOCA/salt-induced hypertension in female rats. J Neuroinflammation. 2015;12:47. https://doi.org/10.1186/s12974-015-0261-4.
Xue B, Zhang Z, Johnson RF, Johnson AK. Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension (Dallas, Tex : 1979). 2012;59(2):459–66. https://doi.org/10.1161/HYPERTENSIONAHA.111.185116.
Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension (Dallas, Tex : 1979). 1999;33(1 Pt 2):323–8.
Dean SA, Tan J, O'Brien ER, Leenen FH. 17beta-estradiol downregulates tissue angiotensin-converting enzyme and ANG II type 1 receptor in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;288(3):R759–66. https://doi.org/10.1152/ajpregu.00595.2004.
Feng Y, Xia H, Santos RA, Speth R, Lazartigues E. Angiotensin-converting enzyme 2: A new target for neurogenic hypertension. Exp Physiol. 2010;95(5):601–6. https://doi.org/10.1113/expphysiol.2009.047407.
Xue B, Badaue-Passos D Jr, Guo F, Gomez-Sanchez CE, Hay M, Johnson AK. Sex differences and central protective effect of 17beta-estradiol in the development of aldosterone/NaCl-induced hypertension. Am J Physiol Heart Circ Physiol. 2009;296(5):H1577–85. https://doi.org/10.1152/ajpheart.01255.2008.
Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor-alpha mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol. 2007;292(4):H1770–6. https://doi.org/10.1152/ajpheart.01011.2005.
Saleh TM, Connell BJ, Saleh MC. Acute injection of 17beta-estradiol enhances cardiovascular reflexes and autonomic tone in ovariectomized female rats. Auton Neurosci. 2000;84(1–2):78–88.
Xue B, Singh M, Guo F, Hay M, Johnson AK. Protective actions of estrogen on angiotensin II-induced hypertension: Role of central nitric oxide. Am J Physiol Heart Circ Physiol. 2009;297(5):H1638–46. https://doi.org/10.1152/ajpheart.00502.2009.
Xue B, Zhao Y, Johnson AK, Hay M. Central estrogen inhibition of angiotensin II-induced hypertension in male mice and the role of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2008;295(3):H1025–H32. https://doi.org/10.1152/ajpheart.00021.2008.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Zucker reports grants from NIH, during the conduct of the study. Drs. De Morais and Shanks declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Secondary Hypertension: Nervous System Mechanisms
Rights and permissions
About this article

Cite this article
de Morais, S.D.B., Shanks, J. & Zucker, I.H. Integrative Physiological Aspects of Brain RAS in Hypertension. Curr Hypertens Rep 20, 10 (2018). https://doi.org/10.1007/s11906-018-0810-1
Published:
DOI: https://doi.org/10.1007/s11906-018-0810-1