
Abstract
Purpose of Review
Despite their high prevalence, the pathophysiology of allergic rhinitis (AR) and chronic rhinosinusitis (CRS) remains unclear. Recently, transient receptor potential (TRP) cation channels emerged as important players in type 2 upper airway inflammatory disorders. In this review, we aim to discuss known and yet to be explored roles of TRP channels in the pathophysiology of AR and CRS with nasal polyps.
Recent Findings
TRP channels participate in a plethora of cellular functions and are expressed on T cells, mast cells, respiratory epithelial cells, and sensory neurons of the upper airways. In chronic upper airway inflammation, TRP vanilloid 1 is mostly studied in relation to nasal hyperreactivity. Several other TRP channels such as TRP vanilloid 4, TRP ankyrin 1, TRP melastatin channels, and TRP canonical channels also have important functions, rendering them potential targets for therapy.
Summary
The role of TRP channels in type 2 inflammatory upper airway diseases is steadily being uncovered and increasingly recognized. Modulation of TRP channels may offer therapeutic perspectives.
Similar content being viewed by others
Introduction
Rhinitis and rhinosinusitis represent two clinical entities of chronic upper airway diseases, characterized by inflammation of the (para)nasal mucosa that results in classic pathological features such as rhinorrhea, post-nasal drip, nasal obstruction, nasal itch, sneezing, loss of smell, and/or facial pain and pressure. Based on patient history and clinical findings, i.e., symptom severity, atopy, presence/absence of nasal polyps, and various comorbidities, different phenotypes of rhinitis and rhinosinusitis are defined. Additionally, the underlying mechanisms, or endotypes, are diverse and even overlapping, making it sometimes difficult to clearly distinct different phenotypes [1•].
Considering rhinitis, three major phenotypes can be recognized and distinguished: allergic rhinitis (AR), infectious rhinitis, and the heterogenous group of non-allergic rhinitis (NAR) [2]. In case of chronic rhinosinusitis (CRS), a phenotype with (CRSwNP) and without nasal polyps (CRSsNP) can be distinguished [3••]. In Europe, both AR and CRSwNP feature mainly type 2 inflammatory processes.
Nasal congestion, edema, mucus production, and—in the case of CRSwNP—nasal polyp formation lead to nasal obstruction, whereas rhinorrhea and postnasal drip are the result of mucus production and often impaired mucociliary clearance [4,5,6]. In AR, allergens can cause nasal itch and sneezing through binding of histamine to its receptor on afferent neurons in the superficial mucosal layer [7]. In CRSwNP, nasal congestion impairs the pressure-equalizing properties of the ostia to the paranasal sinuses, resulting in facial pain [8]. Lastly, loss of smell is due to inflammation of the olfactory cleft, in addition to the conductive component caused by nasal obstruction [8].
In Europe, up to 30% and over 10% of the general population suffers from AR [9] and CRS respectively [10], which comes with an enormous socio-economic impact [11,12,13,14]. Despite the high prevalence, a complete understanding of the underlying pathophysiology is missing.
Both AR and CRSwNP share many similarities and are mainly driven by type 2 inflammation (Figs. 1 and 2) [1•]. Indeed, activated Th2 cells produce interleukin 4 (IL-4), IL-5, and IL-13, leading to IgE production and eosinophil recruitment [3, 4]. Additionally, these patients often experience increased leads of sensory neurons to commonly encountered environmental stimuli, including smoke or temperature changes, which leads to neurogenic inflammation. The latter is often referred to as nasal hyperreactivity (NHR) [15••, 16, 17]. Lastly, a defective epithelial barrier is found, due to tight junction defects in addition to altered mucociliary function, which facilitates mucosal penetration of allergens and noxious substances [18•, 19].

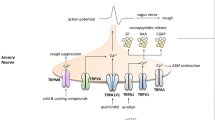
Proposed model of the pathophysiology of allergic rhinitis and the potential role of TRP channels. Allergens presented by dendritic cells induce maturation of Th0 cells to Th2 cells in lymph nodes. Pro-inflammatory mediators such as IL-4, IL-5, and IL-13 are released by Th2 cells and activated type 2 innate lymphoid cells and induce eosinophil recruitment/activation and production of monoclonal allergen-specific IgE. In sensitized individuals, mast cell mediators are released upon binding of allergens to allergen-specific IgE and induce mucus production, vasodilation, and plasma extravasation leading to nasal congestion and edema, ultimately resulting in nasal obstruction, rhinorrhea, and post-nasal drip. On the other hand, epithelial mediators can activate dendritic cells and type 2 innate lymphoid cells. Activation of sensory afferent neurons by histamine results in nasal itch and sneezing. Upon neuronal activation by endogenous or exogenous triggers, the signal travels to the central nervous system, inducing parasympathetic (increased mucus production and vasodilation) or orthosympathetic (vasoconstriction) responses (orthodromic pathway). However, in case of nasal hyperreactivity, neuropeptides are released directly from afferent nerves (antidromic pathway). Lastly, not only environmental factors, but also inflammatory mediators disrupt the epithelial barrier function, increasing epithelial permeability for potentially noxious stimuli. The role of TRPV1, TRPA1, and TRPM8 in neurogenic inflammation has been studied the most. However, TRP channels present on other cells could be potential therapeutic targets as well (gray). Ach, acetylcholine; NA, noradrenaline; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; NMU, neuromedin U; SP, substance P; ILC2, type 2 innate lymphoid cell; PG, prostaglandins; LT, leukotrienes; LN, lymph node; IL, interleukin; LPS, lipopolysaccharide; TSLP, thymic stromal lymphopoietin

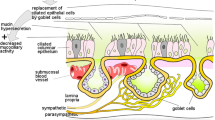
Proposed model of the pathophysiology of chronic rhinosinusitis with nasal polyps and the potential role of TRP channels. Antigens presented by dendritic cells initiate development of Th2 cells that release IL-4, IL-5, and IL-13. IL-5 recruits and activates eosinophils, which ultimately leads to fibrin cross-linking and polyp formation. IL-4 and IL-13 stimulate B cells to produce not only monoclonal IgE to Staphylococcus aureus antigens but also polyclonal autoantibodies leading to complement activation at the basement membrane of the epithelium. Together with protease activity from allergens, environmental factors, and inflammatory mediators, this leads to disruption of the epithelial barrier, facilitating penetration of environmental stimuli. Activated epithelial cells release pro-inflammatory mediators, which can activate dendritic cells and type 2 innate lymphoid cells. Much like in AR, the nervous system participates in the inflammatory process through neuro-immune interactions, modulating the immunological battlefield. Type 2 inflammatory mediators can activate and potentially sensitize sensory afferent neurons, while neuropeptides can activate mast cells and type 2 innate lymphoid cells (ILC2). Moreover, central reflexes regulate vascular tone and mucosal gland activity. The role of TRPV1, TRPA1, and TRPM8 in neurogenic inflammation has been studied the most. However, TRP channels present on other cells could be potential therapeutic targets as well (gray). Ach, acetylcholine; NA, noradrenaline; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; NMU, neuromedin U; SP, substance P; ILC2, type 2 innate lymphoid cell; PG, prostaglandins; LT, leukotrienes; LN, lymph node; IL, interleukin; LPS, lipopolysaccharide; TSLP, thymic stromal lymphopoietin; CCL23, chemokine (C-C motif) ligand 23; FXIIIa, activated coagulation factor 13; t-PA, tissue plasminogen activator
In the last decades, transient receptor potential (TRP) cation channels emerged in the field of upper airway inflammation as active players in the context of NHR, which is present in the majority of patients with rhinitis, CRS, and asthma [17, 20]. TRP channels are implicated in a plethora of physiological processes taking place in the upper airways, including chemosensation [21], thermosensation [22, 23], nociception [24,25,26], regulation of the tone [27, 28] and permeability [29] of the vasculature, release of neuropeptides [15••] and immune cell mediators [30], ciliary beating [31], mucus secretion [32, 33], and barrier function [34]. Considering that every single one of these processes is affected in the context of type 2 inflammation, we find it relevant to explore their potential roles in the pathophysiology of AR and CRSwNP. In this review, we discuss currently known and yet to be explored pathophysiological roles of TRP channels in the related upper airways disease conditions. In particular, we focus on the function of TRP channels in cell types relevant in type 2 upper airway diseases, on how these receptors can influence the pathophysiology of AR and CRS, and on how they may be targeted by novel therapeutic approaches to alleviate patient symptoms.
The TRP Channel Family
Since the first mammalian Trp gene was discovered in 1989 [35], TRP channels gained significant attention, and quickly, it became clear that they are expressed in virtually all tissues throughout the body. TRP channels are largely conserved across species [36], and based on their amino acid sequence homology, six subfamilies of mammalian TRP proteins comprising a total of 28 members have been identified so far: TRPC (canonical 1–7), TRPV (vanilloid 1–6), TRPM (melastatin 1–8), TRPA (ankyrin 1), TRPP (polycystic 1–3), and TRPML (mucolipin 1–3) [36, 37]. TRP channels are built by homo- or hetero-arrangements [36, 38] of four monomers, each with 6 putative transmembrane segments, with the C- and N-termini located in the cytoplasm [39]. The selectivity filter of a TRP channel pore is formed by a re-entering extracellular loop between transmembrane segments 5 and 6 [36, 40]. TRP channels are part of signaling pathways downstream of G protein-coupled receptor activation or activation of cell-specific receptors such as T cell receptors [41,42,43,44,45]. Besides being present on type 2 inflammatory cells, several TRP channels are expressed in sensory neurons and/or epithelial cells (e.g., TRPV1, TRPV2, TRPV3, TRPV4, TRPA1, TRPM2, TRPM3, and TRPM8), where they can be directly activated by mechanical and thermal stimuli, by a wide variety of potentially noxious exogenous chemicals, and by endogenous molecules that signal tissue damage [27, 37, 38, 44, 46,47,48,49,50•].
TRP channel activation, i.e., the transition from a closed to open pore conformation, leads to cation entry at physiological resting membrane potentials, resulting in the increase of intracellular Na+ and Ca2+ concentrations and therefore membrane depolarization [38]. TRP channel activation, inhibition, sensitization, or desensitization can occur in different ways and to different extents, depending on the composition of the tetramer structure, on the applied stimulus, and on post-translational modifications such as phosphorylation [45].
In the upper airways, TRPV1 and TRPA1 are studied the most. They have been reported to be expressed in T cells, mast cells, and afferent neurons and can be activated directly (by environmental triggers) or indirectly (via activation of G protein-coupled receptors or receptor tyrosine kinases) [51••]. Activation of G protein-coupled receptors or receptor tyrosine kinases stimulates phospholipase C, which hydrolyzes phosphatidylinositol-4,5-biphosphate (PIP2) into diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) [51••, 52]. This relieves TRPV1 and TRPA1 from inhibition by PIP2 [51••, 52]. Furthermore, DAG directly not only activates TRPV1 and TRPA1, but it also activates protein kinase C, which on its turn enhances TRP activity [51••, 52]. Also, G protein-coupled receptors can activate phospholipase A2, generating arachidonic acid of which the metabolites, such as 12-hydroxyperoxyeicosatetraenoic acid or prostaglandin E2, can activate TRP channels [51••, 52]. Lastly, since TRP channels are Ca2+-permeable, they could play a role in the phenomenon of store-operated Ca2+ entry (SOCE) [53]. After hydrolysis of PIP2 into DAG and IP3, the latter binds on its receptor located on the endoplasmic reticulum, leading to release of Ca2+ from this intracellular store [51••, 53]. Next, stromal interaction-molecule 1 activates Ca2+-release activated channels (CRAC) formed by Orai1 subunits leading to a consistent Ca2+ influx and store-operated Ca2+ channels (SOC) formed by Orai1 and TRPC proteins resulting in non-selective cation influx [41, 53].
TRP Channels in Type 2 Upper Airway Inflammation
T Cells
Ca2+ plays an important role in intracellular signaling and cellular responses in T cells [41, 45, 54]. Cytokine secretion and T cell proliferation depend on increased intracellular Ca2+ concentration [43]. Generally, after activation of the T cell receptor, phospholipase C is activated leading to an initial Ca2+ influx from the extracellular environment and intracellular stores. Subsequent activation of CRAC results in a sustained Ca2+ influx [41, 45, 55]. Increased body temperature during inflammation or infection enhances immune responses, suggesting involvement of thermoregulated ion channels such as TRP channels in immunologic processes [56, 57]. On CD4+ T cells, there is evidence of expression of TRPA1, TRPC1/2/3/5, TRPV1/2/3/4, and TRPM1/2/4/5/6/7 (Table 1) [45].
TRPA1 expression increases after TCR activation and correlates with T cell activation [54]. As hypothesized by Bertin et al., TRPA1 and TRPV1 subunits together can form heteromeric channels, resulting in TRPV1-hyperactivation in Trpa1 knockout cells due to formation of solely TRPV1 homomers with higher functionality [58].
Upon TCR activation, lymphocyte-specific protein tyrosine kinase phosphorylates TRPV1, regulating its activity [55]. Phosphorylation by lymphocyte-specific protein tyrosine kinase leads to direct activation, sensitization for endogenous agonists such as DAG and recruitment of TRPV1 channels from intracellular pools to the cell membrane [45]. Also, upon TCR activation, TRPV1 and TRPV4 expressions on T cells are upregulated and TRPV1 rapidly migrates towards TCR clusters [55, 57].
TCR-mediated activation induces release of adenosine 5′-diphosphoribose (ADPR) from the endoplasmic reticulum, which subsequently activates TRPM2 channels [80]. Furthermore, murine TRPM2 seems to facilitate T cell proliferation and mediate secretion of pro-inflammatory cytokines [45]. TRPM2 is suspected to maintain T cell function in an inflammatory milieu [62]. Activation of TRPM4, a monovalent cation-permeable (but Ca2+-impermeable) channel, depolarizes the plasma membrane, hence reducing the driving force for SOCE-mediated Ca2+-entry, preventing intracellular Ca2+-overload [63, 81].
Lastly, as stated above, TRPC1 and TRPC3 are involved in SOCE, although their precise roles remain to be clarified [45, 82]. In mice, TRPC5 is suspected to mediate the regulatory T cell–induced decrease of effector T cell activity [45].
Mast Cells
TRP channels expressed on mast cells include TRPC1–7, TRPV1/2/4/6, TRPM2/4/7/8, and TRPA1 [42]. In these cells, Ca2+ is involved in activation of transcription factors, as well as in the production and release of mast cell mediators [41, 42]. The cross-linking of the high-affinity receptor FcεRI results in activation of phospholipase C with subsequent Ca2+ mobilization into the cytoplasm from the extracellular space and from intracellular stores, followed by SOCE [42, 65]. Depending on the spatial and temporal pattern of the increase of the intracellular Ca2+ concentration, different cellular responses are triggered, such as degranulation and chemotaxis [42]. Ca2+ influx triggers the release of mast cell mediators such as histamine and eicosanoids to the extracellular environment [83].
In resting mast cells, TRPA1 is localized on intracellular vesicles, where it can interact with secretogranin III, and thus, it may play a role in formation of mast cell granules [84]. Also, TRPA1 has been shown to play a role in hypoxia-induced mast cell degranulation [85].
TRPV1 stimulation with its specific agonist capsaicin results in an inward Ca2+ current, but this does not lead to mast cell degranulation [86]. In contrast, TRPV2 activation by physical triggers, such as mechanical stress, very high temperatures, and light of 640 nm, does induce mast cell degranulation [87, 66].
TRPM4 is rapidly activated after FcεRI stimulation, depolarizing mast cells and hence counter-acting SOCE, similar to the situation in T cells [30]. TRPM4 seems to be involved also in Ca2+-dependent mast cell migration [88].
Lastly, TRPC channels are activated after G protein-coupled receptor or receptor tyrosine kinase stimulation, but are also involved in SOCE [42, 82]. Like in T cells, their exact roles here need further clarification, but TRPC5 seems to associate with STIM1 and Orai1, forming Ca2+-permeable SOC channels [89].
Upper Respiratory Epithelial Cells
TRP channels are expressed in epithelial cells of the nasal and paranasal mucosa, but their functional roles remain to be fully determined. Immunohistochemistry of the nasal mucosa showed expression of TRPV1–4 around seromucinous glands and capillary endothelial cells in the lamina propria, as well as in epithelial cells [90]. In a study from Bhargave et al., the expression of TRPV1/2/4/6 and TRPC1/3/4/6 was found in sinonasal mucosal biopsies using RT-q-PCR [91]. No TRPV4 expression was found on ciliated epithelial cells using immunohistochemistry. However, commercial antibodies for TRP channels are often poorly validated. For instance, in the latter paper, rabbit anti-mouse TRPV4 antibodies were used on human tissue [42, 91]. Recently, TRPV4 was found to be functionally expressed in nasal epithelial cells [31, 75].
Stimulation of the menthol- and cold-activated TRPM8 in sensory neurons induces a subjectively enhanced nasal patency without any objective ground [76, 92]. Contrastingly, menthol and cold air induce an increase in nasal secretions [76]. Indeed, activation of TRPM8 on nasal epithelial cells upregulates MUC5AC at both mRNA and protein level [76]. Nasal challenges with capsaicin and allyl isothiocyanate (a TRPA1 agonist) produced an increase in MUC5B in nasal lavage fluid, though the underlying mechanism remains to be clarified [93].
Trigeminal Sensory Neurons
In trigeminal neurons, TRP channels are highly expressed in unmyelinated sensory C fibers and are suspected to play key roles in the phenomenon of NHR [15••]. TRP channels in these cells have been proposed to be of importance in neuro-immune interactions [94]. This may take place due to their sensory roles in the context of infection (e.g., by sensing LPS [95,96,97]) or through the detection of exogenous noxious chemicals or endogenous molecules signaling tissue damage [21, 98]. Most interest has been put in TRPV1 and TRPA1, which often co-localize with neuropeptides such as substance P (SP), calcitonin gene-related peptide (CGRP), and neurokinin A (NKA) [99,100,101]. NHR is suspected to be caused by overactivity or sensitization of these TRP channels on sensory afferent neurons, leading to release of neuropeptides in the mucosal and submucosal space, triggering vasodilation and mucus secretion [15••]. Studies on trigeminal neurons are scarce, but data on dorsal root ganglion neurons projecting to the lower airways indicate release of SP, CGRP, and NKA upon activation of TRPV1 or TRPA1 [102,103,104,105]. Likewise, oromucosal induce of capsaicin - which activates TRPV1 - or menthol - which can activate both TRPM8 and TRPA1 [106] - induce release of CGRP [107]. Lastly, immunohistochemistry studies and PCR showed presence of TRPV2, TRPM3, TRPC1, TRPC3, and TRPC4 in trigeminal ganglionic neurons of lab animals [108,109,110,111,112].
TRP Channels as a Potential Therapeutic Target in Type 2 Inflammation of the (Sino-)Nasal Mucosa
Studies on TRP channels in upper airway inflammation have focused mainly on idiopathic rhinitis patients, where an overexpression of TRPV1 and increased levels of SP in nasal secretions were linked to neurogenic inflammation and NHR [15••]. However, it is progressively becoming clear that TRP channels are implicated in other underlying pathophysiological mechanisms as well. Inflammatory mediators found to be increased in nasal secretions of patients with type 2 upper airway inflammation are known to sensitize various TRP channels [51••]. For example, nerve growth factor is reported to be increased in nasal secretions and blood of AR patients and CRSwNP patients [46, 113, 114]. In the nose, nerve growth factor secreted by eosinophils can sensitize TRPV1 and TRPA1 in sensory nerve endings, increase SP content of sensory neurons, and induce dendrite sprouting [5, 51••, 114,115,116]. In allergic inflammation, bradykinin induces plasma extravasation and vasodilation [117]. Increasing evidence points towards sensitizing properties of bradykinin for TRPV1, TRPV4, and TRPA1 [51••, 118, 119]. At last, prostaglandin E2, which skews towards a type 2 inflammation and yields anti-inflammatory properties [120], sensitizes TRPV1 [51••, 121] and activates TRPA1 via its electrophilic metabolites [122, 123]. In the following, we discuss previously described and potential roles of TRP channels in type 2 inflammation of the upper airways.
TRPV1
As for idiopathic rhinitis, up to two-thirds of AR and CRS patients report NHR [15••, 17, 124]. Studies on idiopathic rhinitis patients indicate a role of the TRPV1-SP pathway, leading to neurogenic inflammation [15••]. Here, the release of SP—but possibly also other neuromediators such as CGRP, NKA, or neuromedin U—leads to vasodilation and increased mucus secretion. Whether the same mechanism is present in AR and CRS and how it is influenced by immunologic mediators has to be investigated.
Considering TRPV1 in AR, several observations are interesting. For instance, one study found TRPV1 expression based on RT-q-PCR to be significantly decreased in nasal mucosa of AR and NAR patients [125], whereas another described an increased expression of TRPV1 in idiopathic rhinitis patients, a specific subgroup of NAR [72]. Studies on seasonal AR report that more symptoms and pain are induced when stimulating TRPV1 intranasally with capsaicin during the allergy season compared with challenges outside of the season, suggesting sensitization to capsaicin during allergic inflammation [126,127,128]. Capsaicin treatment in AR patients did not lower total nasal symptom scores but decreased their sensitivity to histamine challenges [129]. Azelastine, originally developed as an antihistaminic drug, desensitizes TRPV1, partly by internalization of the channel from the plasma membrane [71]. In combination with fluticasone, it decreases nasal symptoms, nasal hyperreactivity, and SP levels in nasal secretions of perennial AR patients [130]. Lastly, sole inhibition of TRPV1 did not reduce symptoms triggered by allergen challenge, nor did it affect the total nasal symptom score in seasonal AR patients [131, 132]. Hence, inhibition of TRPV1 could be useful in targeting neurogenic inflammation but seems to lack a direct effect on allergic inflammation. After all, the pathophysiology of AR is more complex than the purely neurogenic inflammation present in idiopathic rhinitis patients, and symptoms are also induced via type 2 inflammatory mechanisms. Since NHR has also been reported in AR, these patients might benefit from capsaicin therapy [72, 133•], targeting the neurogenic component of the pathophysiological pathway. Of particular interest is that the capsaicin treatment reduced the hyperreactivity to the model noxious chemical allyl isothiocyanate [16], which could be partly mediated by activation of TRPV1 in sensory neurons [134,135,136]. On the other hand, Trpv1 knockout mice, as well as mice treated with a pharmacological antagonist of TRPV1, exhibited less type 2 inflammatory mediators after allergic sensitization [59]. Further research on the role of TRPV1 and the therapeutic potential of capsaicin or TRPV1-inhibitors in AR is needed.
In CRSwNP patients, one study found decreased TRPV1 and TRPA1 mRNA levels in nasal mucosa, whereas TRPV1 was upregulated in case of comorbid asthma or allergy [137]. Another research group described more TRPV1 mRNA in nasal lavage fluid of CRS patients compared with healthy controls but did not investigate the atopic status [138]. Hence, it could be that TRPV1 expression depends on the presence of comorbid allergy.
Lastly, the activation of upper respiratory epithelial TRPV1 enhances the release of pro-inflammatory mediators [71, 73, 74] and nasal epithelial expression of TRPV1 was recently shown to be increased in asthmatic patients [74]. TRPV1 is shown to mediate acidity-induced barrier dysfunction in the lower airways [139]. Hence, overexpression or overactivity of epithelial TRPV1 might contribute to the defective barrier function in both AR and CRSwNP [18•, 19, 139].
In summary, TRPV1 is mostly associated with NHR and neurogenic inflammation. However, it also facilitates T cell activation with consequent release of inflammatory mediators, playing a role very early in the pathophysiological pathway of type 2 inflammation. In addition, the activation of TRPV1 in upper respiratory epithelial cells could enhance the inflammatory process. Hence, TRPV1 is implicated in all aspects of the pathophysiological mechanisms underlying AR and CRSwNP and leading to nasal symptoms, rendering it a worthy candidate for pharmacological modulation.
TRPA1
Despite the scarcity of studies on TRPA1 in specific inflammatory disorders of the upper airways, this TRP channel deserves to be mentioned. TRPA1 is expressed in sensory afferent neurons in both upper and lower airways and can be activated by a plethora of noxious exogenous stimuli [105, 140]. For example, environmental pollutants, diesel exhaust particles, and compounds within cigarettes smoke induce nociceptive neuronal activation via TRPA1 [140]. On the other hand, endogenous inflammatory mediators such as bradykinin and prostaglandins trigger protein kinase C-mediated activation of TRPV1 and TRPA1 [141]. Lastly, TRPA1 is often put front as a sensor for tissue damage [98, 105] inducing defensive host reflexes. Indeed, TRPA1-mediated activation of perivascular sensory nerves induces vasodilation, contributing to nasal obstruction and hence protecting the lower airways from potentially harmful triggers [142].
Some studies in the lower airways found TRPA1 in immune cells and in airway epithelial cells [140]. Here, a bi-directional process takes place, where activation of TRPA1 leads to a release of inflammatory mediators, while the latter can facilitate the trafficking of TRPA1 to the cell membrane [140].
As mentioned earlier, NHR is a key feature of rhinitis and of rhinosinusitis. NHR is currently best diagnosed with a cold, dry air provocation test [143], and TRPA1 is activated by cold temperatures [144]. In rhinitis patients, TRPA1 mRNA levels are increased [125] and a decreased threshold for neuronal activation in response to the TRPA1 agonist allyl isothiocyanate was observed [16]. Besides, after treatment with a fixed combination of fluticasone and azelastine, which induces chemical desensitization of neurons co-expressing TRPV1 and TRPA1, symptom scores improved in patients with perennial AR [130]. Studies focusing specifically on TRPA1-blockade are needed, both for exploration of therapeutic horizons, as well as to clarify the functional role of TRPA1 in upper respiratory diseases.
TRPV4
TRPV4 is ubiquitously expressed and can be activated by a variety of both physical and chemical stimuli, including hypotonicity, high temperature, and metabolites of arachidonic acid [145]. It mediates release of pro-inflammatory cytokines from T cells [57] and is overexpressed in CRS patients [91]. Hence, TRPV4 potentially enhances or maintains the inflammatory process, suggesting therapeutic possibilities for TRPV4 antagonism. On the other hand, activation of epithelial TRPV4 increases ciliary beat frequency [31, 75], enhancing an often impaired mucociliary clearance [6]. Activation of TRPV4 by LPS leads to a TLR4-independent increase in intracellular Ca2+ concentration, which in turn triggers the release of nitric oxide and an increase in the ciliary beat frequency. Hence, it was postulated that TRPV4 plays a role in protective responses to gram-negative bacterial infections [31]. This may suggest TRPV4 stimulation as potential therapeutic mechanism. However, the effects of TRPV4 stimulation or inhibition in human upper airway diseases remain to be studied.
TRPV4 is currently being investigated as a potential therapeutic target in cough arising from the lower airways, pulmonary edema, and other respiratory and non-respiratory diseases [146, 147]. Due to its abundant expression throughout the body, systemic administration might lead to unexpected side effects [146]. For upper airway diseases, this could easily be dealt with by administering drugs via a nasal spray directly on the nasal mucosa, limiting systemic absorption.
TRPM Channels
Activation of respiratory epithelial TRPM8, for example by menthol or by cooling, is suspected to lead to increased mucus production [76]. This likely results in rhinorrhea, postnasal drip, and nasal obstruction, key symptoms of upper airway inflammation. Also, TRPM8 activation increases the release of pro-inflammatory cytokines by T cells [60], possibly leading to a broad variety of nasal symptoms. The inhibition of TRPM8 could therefore have both a direct effect on nasal symptoms (rhinorrhea, postnasal drip, nasal obstruction), as well as an indirect effect via reduced type 2 inflammatory activity. Based on immunohistochemistry data however, TRPM8 is equally expressed and distributed in nasal mucosa of healthy controls and AR and NAR patients [76, 148, 149]. Further studies are needed to explore the therapeutic perspectives of TRPM8-modulation.
Given their role in both T cell and mast cell activation, the inhibition of TRPM2 [45, 68] and TRPM7 [69] could be considered as a method to decrease type 2 inflammation. The recent identification of TRPM3 as regulator of the tone of resistance arteries [150] suggests an opportunity to target the pathological vasodilation in the upper airways that reduces nasal patency [150]. On the other hand, TRPM4 activation reduces the electrical driving force for Ca2+ to enter the cell, possibly reducing pro-inflammatory activity of T cells [63, 81, 64] and mast cells [30]. Therefore, patients with AR or CRSwNP could possibly benefit from drugs stimulating or sensitizing TRPM4, hence reducing inflammation.
TRPC5/6
TRPC6 is overexpressed on epithelial cells of CRSwNP patients, and its activation induces production of IL-1ß, IL-5, and IL-25 [151]. TRPC5 is upregulated in polyp tissue [152] and mediates regulatory T cell-induced reduction of the pro-inflammatory T helper cell activity [45]. This potentially counteracts excessive inflammatory responses, just as increased expression of TRPV4 in CRS patients possibly is a counter mechanism that enhances impaired mucociliary clearance. Targeting TRPC5 with agonists could be a way to temper inflammation by stimulating the body’s own control mechanisms.
Conclusions
TRP channels are widely expressed, conserved cation channels that regulate intracellular Ca2+-dependent signaling pathways and contribute to a plethora of cellular processes. In the last decades, their cellular functions and their roles in multicellular processes and interactions have been studied intensively. In type 2 upper airway inflammatory disorders, the battlefield is mainly occupied by T cells, mast cells, epithelial cells, and sensory neurons, each releasing their respective mediators and hence interacting with each other, influenced by neuro-immune interactions and barrier defects. The role of TRP channels in these diseases is only starting to be clarified. Even though potential therapeutic targets have already emerged, a TRiP to successful treatment of type 2 upper airway inflammation awaits.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• De Greve G, Hellings PW, Fokkens WJ, Pugin B, Steelant B, Seys SF. Endotype-driven treatment in chronic upper airway diseases. Clin Transl Allergy. 2017;7(22). Concise review covering the importance of endotyping and phenotyping in rhinitis and rhinosinusitis.
Hellings PW, Klimek L, Cingi C, Agache I, Akdis C, Bachert C, et al. Non-allergic rhinitis: position paper of the European Academy of Allergy and Clinical Immunology. Allergy. 2017;72(11):1657–65.
•• Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. 2020;58(S29):1–464. Hallmark paper on the pathophysiology and diagnostic and therapeutic approaches towards rhinosinusitis.
Sin B, Togias A. Pathophysiology of allergic and nonallergic rhinitis. Proc Am Thorac Soc. 2011;8:106–14.
Sarin S, Undem B, Sanico A, Togias A. The role of the nervous system in rhinitis. J Allergy Clin Immunol. 2006;118(5):999–1014.
Chen B, Shaari J, Claire SE, Palmer JN, Chiu AG, Kennedy DW, et al. Altered sinonasal ciliary dynamics in chronic rhinosinusitis. Am J Rhinol. 2006;20(3):325–9.
Pfaar O, Raap U, Holz M, Hörmann K, Klimek L. Pathophysiology of itching and sneezing in allergic rhinitis. Swiss Med Wkly. 2009;139(3–4):35–40.
Eccles R. Mechanisms of the symptoms of rhinosinusitis. Rhinology. 2011;49(2):131–8.
Bousquet J, Khaltaev N, Cruz AA, Denburg J, Fokkens WJ, Togias A, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008. Allergy. 2008;63(s86):8–160.
Hastan D, Fokkens WJ, Bachert C, Newson RB, Bislimovska J, Bockelbrink A, et al. Chronic rhinosinusitis in Europe - an underestimated disease. A GA2LEN study. Allergy Eur J Allergy Clin Immunol. 2011;66(9):1216–23.
De La Hoz CB, Rodríguez M, Fraj J, Cerecedo I, Antolín-Amérigo D, Colás C. Allergic rhinitis and its impact on work productivity in primary care practice and a comparison with other common diseases: the Cross-sectional study to evAluate work Productivity in allergic Rhinitis compared with other common dIseases (CAPRI) study. Am J Rhinol Allergy. 2012;26(5):390–4.
Blaiss MS, Hammerby E, Robinson S, Kennedy-martin T, Buchs S. The burden of allergic rhinitis and allergic rhinoconjunctivitis on adolescents: a literature review. Ann Allergy Asthma Immunol. 2018;121(1):43–52.
Colás C, Brosa M, Antón E, Montoro J, Navarro A, Dordal MT, et al. Estimate of the total costs of allergic rhinitis in specialized care based on real-world data: the FERIN Study. Allergy Eur J Allergy Clin Immunol. 2017;72(6):959–66.
Lourijsen ES, Fokkens WJ, Reitsma S. Direct and indirect costs of adult patients with chronic rhinosinusitis with nasal polyps. Rhinology. 2020;58(3):213–7.
•• Van Gerven L, Steelant B, Hellings PW. Nasal hyperreactivity in rhinitis: a diagnostic and therapeutic challenge. Allergy. 2018;73(9):1784–91. State-of-the-art on the phenomenon of nasal hyperreactivity.
Van Gerven L, Alpizar YA, Steelant B, Callebaut I, Kortekaas Krohn I, Wouters M, et al. Enhanced chemosensory sensitivity in patients with idiopathic rhinitis and its reversal by nasal capsaicin treatment. J Allergy Clin Immunol. 2017;140(2):437–46.
Doulaptsi M, Steelant B, Prokopakis E, Ierodiakonou D, Tsinaslanidou Z, Cools L, et al. Prevalence and impact of nasal hyperreactivity in chronic rhinosinusitis. Allergy. 2020;12:1–4.
• Steelant B, Seys SF, Van Gerven L, Van Woensel M, Farré R, Wawrzyniak P, et al. Histamine and T helper cytokine–driven epithelial barrier dysfunction in allergic rhinitis. J Allergy Clin Immunol. 2018;141(3):951–63. Interesting research on the interaction between type 2 inflammatory cells and barrier function in allergic rhinitis.
Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol. 2012 Nov;130(5):1087–1096.e10.
Feijen J, Seys SF, Steelant B, Bullens DMA, Dupont LJ, García-Cruz M, et al. Prevalence and triggers of self-reported nasal hyperreactivity in adults with asthma. World Allergy Organ J. 2020;13(6):100132.
Roper SD. TRPs in taste and Chemesthesis. Handb Exp Pharmacol. 2014;223:827–71.
Mori Y, Voets T. Sensors and regulatory mechanisms of thermal physiology. Pflugers Arch Eur J Physiol. 2018;470(5):703–4.
Vriens J, Nilius B, Voets T. Peripheral thermosensation in mammals. Nat Rev Neurosci. 2014;15(9):573–89.
Hung C-Y, Tan C-H. TRP channels in nociception and pathological pain. In: Advances in pain research: mechanisms and modulation of chronic pain. 2018. p. 13–27.
González-Ramírez R, Chen Y, Liedtke WB, Morales-Lázaro SL. TRP channels and pain. In: Neurobiology of TRP channels. 2019. p. 125–48.
Mickle AD, Shepherd AJ, Mohapatra DP. Nociceptive TRP channels: sensory detectors and transducers in multiple pain pathologies. Pharmaceuticals. 2016;9(4):72.
Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev. 2015;95(2):645–90.
Alonso-Carbajo L, Kecskes M, Jacobs G, Pironet A, Syam N, Talavera K, et al. Muscling in on TRP channels in vascular smooth muscle cells and cardiomyocytes. Cell Calcium. 2017;66:48–61.
Genova T, Gaglioti D, Munaron L. Regulation of vessel permeability by TRP channels. Front Physiol. 2020;11(421).
Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, et al. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol. 2007;8(3):312–20.
Alpizar YA, Boonen B, Sanchez A, Jung C, López-Requena A, Naert R, et al. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat Commun. 2017;8(1).
Cantero-Recasens G, Butnaru CM, Brouwers N, Mitrovic S, Valverde MA, Malhotra V. Sodium channel TRPM4 and sodium/calcium exchangers (NCX) cooperate in the control of Ca2-induced mucin secretion from goblet cells. J Biol Chem. 2019;294(3):816–26.
Zholos A. TRP channels in respiratory pathophysiology: the role of oxidative, chemical irritant and temperature stimuli. Curr Neuropharmacol. 2015;13(2):279–91.
Mukaiyama M, Usui T, Nagumo Y. Non-electrophilic TRPA1 agonists, menthol, carvacrol and clotrimazole, open epithelial tight junctions via TRPA1 activation. J Biochem. 2020;ePub ahead of print.
Montell C, Rubin GM. Molecular characterization of the drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2(4):1313–23.
Samanta A, Hughes TET, Moiseenkova-Bell VY. Transient receptor potential (TRP) channels. Subcell Biochem. 2018;87:141–65.
Nilius B. TRP channels in disease. Biochim Biophys Acta - Mol Basis Dis. 2007;1772(8):805–12.
Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. 2010;2(10).
Gaudet R. TRP channels entering the structural era. J Physiol. 2008;586(15):3565–75.
Owsianik G, Talavera K, Voets T, Nilius B. Permeation and selectivity of Trp channels. Annu Rev Physiol. 2006;68:685–717.
Parenti A, De Logu F, Geppetti P, Benemei S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br J Pharmacol. 2016;173(6):953–69.
Freichel M, Almering J, Tsvilovskyy V. The role of TRP proteins in mast cells. Front Immunol. 2012;3(150):1–15.
Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. 2009;10(1):21–7.
Khalil M, Alliger K, Weidinger C, Yerinde C, Wirtz S, Becker C, et al. Functional role of transient receptor potential channels in immune cells and epithelia. Front Immunol. 2018;9(174).
Bertin S, Raz E. Transient receptor potential (TRP) channels in T cells. Semin Immunopathol. 2016;38(3):309–19.
Van Gerven L, Boeckxstaens G, Hellings PW. Up-date on neuro-immune mechanisms involved in allergic and non-allergic rhinitis. Rhinology. 2012;50(3):227–35.
Klein AH. The orotrigeminal system. 1st ed. Vol. 164, Handbook of clinical neurology. Elsevier B.V.; 2019. 205–216 p.
Vangeel L, Benoit M, Miron Y, Miller PE, De Clercq K, Chaltin P, et al. Functional expression and pharmacological modulation of TRPM3 in human sensory neurons. Br J Pharmacol. December 2019;2020:2683–95.
Atsumi K, Yajima T, Tachiya D, Kokubun S, Shoji N, Sasano T, et al. Sensory neurons in the human jugular ganglion. Tissue Cell. 2020;64(February):101344.
• Balemans D, Boeckxstaens GE, Talavera K, Wouters MM. Transient receptor potential ion channel function in sensory transduction and cellular signaling cascades underlying visceral hypersensitivity. Am J Physiol - Gastrointest Liver Physiol. 2017;312(6):G635–48. Important study illustrating the neuro-immune interaction with the mast cell mediator histamine and TRP-channels.
•• Gouin O, L’Herondelle K, Lebonvallet N, Le Gall-Ianotto C, Sakka M, Buhé V, et al. TRPV1 and TRPA1 in cutaneous neurogenic and chronic inflammation: pro-inflammatory response induced by their activation and their sensitization. Protein Cell. 2017;8(9):644–61. Concise overview of intracellular signaling cascades after activation of the most studied TRP channels in the upper airways: TRPV1 and TRPA1.
Rosenbaum T, Simon S a. TRPV1 receptors and signal transduction. In: Liedtke WB, Heller S, editors. TRP ion channel function in sensory transduction and cellular signaling cascades. CRC Press/Taylor & Francis; 2007.
Lopez JJ, Jardin I, Sanchez-Collado J, Salido GM, Smani T, Rosado JA. TRPC channels in the SOCE scenario. Cells. 2020;9(1).
Sahoo SS, Majhi RK, Tiwari A, Acharya T, Kumar PS, Saha S, et al. Transient receptor potential ankyrin1 channel is endogenously expressed in T cells and is involved in immune functions. Biosci Rep. 2019;39(9).
Bertin S, Aoki-nonaka Y, De Jong PR, Nohara LL, Xu H, Stanwood SR, et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4+ T cells. Nat Immunol. 2014;15(11):1055–63.
Smith JB, Knowlton RP, Agarwal SS. Human lymphocyte responses are enhanced by culture at 40 degrees. J Immunol. 1978;121(2):691–4.
Majhi RK, Sahoo SS, Yadav M, Pratheek BM, Chattopadhyay S, Goswami C. Functional expression of TRPV channels in T cells and their implications in immune regulation. FEBS J. 2015;282(14):2661–81.
Bertin S, Aoki-Nonaka Y, Lee J, De Jong PR, Kim P, Han T, et al. The TRPA1 ion channel is expressed in CD4+ t cells and restrains T-cell-mediated colitis through inhibition of TRPV1. Gut. 2017;66(9):1584–96.
Samivel R, Kim DW, Son HR, Rhee Y-H, Kim EH, Kim JH, et al. The role of TRPV1 in the CD4+ T cell-mediated inflammatory response of allergic rhinitis. Oncotarget. 2016;7(1):148–60.
Kume H, Tsukimoto M. TRPM8 channel inhibitor AMTB suppresses murine T-cell activation induced by T-cell receptor stimulation, concanavalin A, or external antigen re-stimulation. Biochem Biophys Res Commun. 2019;509(4):918–24.
Ghoneum MH, Gimzewski JK, Ghoneum A, Katano H, Nila Paw U C, Agrawal A. Inhibition of TRPV1 channel activity in human CD4+ T cells by nanodiamond and nanoplatinum liquid, DPV576. Nanomaterials. 2018;8(10).
Trebak M, Kinet J-P. Calcium signalling in T cells. Nat Rev Immunol. 2019;19(3):154–69.
Wu L, Sweet T, Clapham DE. Current progress in the mammalian TRP ion channel family. Pharm Rev. 2010;62(3):381–404.
Weber KS, Hildner K, Murphy KM, Allen PM. TRPM4 differentially regulates Th1 and Th2 funtion by altering calcium signaling and NFAT localization. J Immunol. 2010;85(5):2836–46.
Lam RS, Shumilina E, Matzner N, Zemtsova IM, Sobiesiak M, Lang C, et al. Phosphatidylinositol-3-kinase regulates mast cell ion channel activity. Cell Physiol Biochem. 2008;22(1–4):169–76.
Stokes AJ, Shimoda LMN, Koblan-Huberson M, Adra CN, Turner H. A TRPV2-PKA signaling module for transduction of physical stimuli in mast cells. J Exp Med. 2004;200(2):137–47.
Yang WZ, Chen JY, Yu JT, Zhou LW. Effects of low power laser irradiation on intracellular calcium and histamine release in RBL-2H3 mast cells. Photochem Photobiol. 2007;83(4):979–84.
Oda S, Uchida K, Wang X, Lee J, Shimada Y, Tominaga M, et al. TRPM2 contributes to antigen-stimulated Ca2+ influx in mucosal mast cells. Pflugers Arch Eur J Physiol. 2013;465(7):1023–30.
Huang L, Ng NM, Chen M, Lin X, Tang T, Cheng H, et al. Inhibition of TRPM7 channels reduces degranulation and release of cytokines in rat bone marrow-derived mast cells. Int J Mol Sci. 2014;15(7):11817–31.
Medic N, Desai A, Komarow H, Burch LH, Bandara G, Beaven MA, et al. Examination of the role of TRPM8 in human mast cell activation and its relevance to the etiology of cold-induced urticaria. Cell Calcium. 2011;50(5):473–80.
Singh U, Bernstein JA, Haar L, Luther K, Jones WK. Azelastine desensitization of transient receptor potential vanilloid 1: a potential mechanism explaining its therapeutic effect in nonallergic rhinitis. Am J Rhinol Allergy. 2014;28(3):215–24.
Van Gerven L, Alpizar YA, Wouters MM, Hox V, Hauben E, Jorissen M, et al. Capsaicin treatment reduces nasal hyperreactivity and transient receptor potential cation channel subfamily V, receptor 1 (TRPV1) overexpression in patients with idiopathic rhinitis. J Allergy Clin Immunol. 2014;133(5):1332–9.
Seki N, Shirasaki H, Kikuchi M, Himi T. Capsaicin induces the production of IL-6 in human upper respiratory epithelial cells. Life Sci. 2007;80(17):1592–7.
Schiffers C, Hristova M, Habiboic A, Dustin CM, Danyal K, Reynaert NL, et al. The transient receptor potential channel vanilloid 1 (TRPV1) is critical in innate airway epithelial responses to protease allergens. Am J Respir Cell Mol Biol. 2020;Epub ahead of print.
Alenmyr L, Uller L, Greiff L, Edward DH. TRPV4-mediated calcium influx and ciliary activity in human native airway epithelial cells. Basic Clin Pharmacol Toxicol. 2014;114(2):210–6.
Liu SC, Lu HH, Fan HC, Wang HW, Chen HK, Lee FP, et al. The identification of the TRPM8 channel on primary culture of human nasal epithelial cells and its response to cooling. Med (United States). 2017;96(31):e7640.
Kageneck C, Nixdorf-bergweiler BE, Messlinger K, Fischer MJM. Release of CGRP from mouse brainstem slices indicates central inhibitory effect of triptans and kynurenate. J Headache Pain. 2014;15:7.
Dux M, Vogler B, Schramm J, Manchen J, Messlinger K. High - dose phenylephrine increases meningeal blood flow through TRPV1 receptor activation and release of calcitonin gene - related peptide. Eur J Pain. 2020;24(2):383–97.
Mckemy DD, Neuhausser WM, Julius D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Vol. 416, Nature. 2002. p. 52–8.
Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, et al. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem. 2006;281(5):2489–96.
Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. TRPM4 regulates calcium oscillations after T cell activation. Science (80- ). 2004;306(5700):1374–7.
Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9(5):399–410.
Ma HT, Beaven MA. Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol. 2011;716:62–90.
Prasad P, Yanagihara AA, Small-Howard AL, Turner H, Stokes AJ. Secretogranin III directs secretory vesicle biogenesis in mast cells in a manner dependent upon interaction with chromogranin A. J Immunol. 2008;181(7):5024–34.
Matsuda K, Okamoto N, Kondo M, Arkwright PD, Karasawa K, Ishizaka S, et al. Mast cell hyperactivity underpins the development of oxygen-induced retinopathy. J Clin Invest. 2017;127(11):3987–4000.
Bíró T, Maurer M, Modarres S, Lewin NE, Brodie C, Ács G, et al. Characterization of functional vanilloid receptors expressed by mast cells. Blood. 1998;91(4):1332–40.
Zhang D, Spielmann A, Wang L, Ding G, Huang F, Gu Q, et al. Mast-cell degranulation induced by physical stimuli involves the activation of transient-receptor-potential channel TRPV2. Physiol Res. 2012;61(1):113–24.
Shimizu T, Owsianik G, Freichel M, Flockerzi V, Nilius B, Vennekens R. TRPM4 regulates migration of mast cells in mice. Cell Calcium. 2009;45(3):226–32.
Ma H-T, Peng Z, Hiragun T, Iwaki S, Gilfillan AM, Beaven MA. Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr 2+ entry, optimal influx of Ca2+ , and degranulation in a rat mast cell line. J Immunol 2008;180(4):2233–2239.
Mohamed Khalifa A, Takumida M, Ishibashi T, Hamamoto T, Hirakawa K. Expression of transient receptor potential vanilloid (TRPV) families 1, 2, 3, and 4 in the mouse olfactory epithelium. Rhinology. 2009;47(3):242–7.
Bhargave G, Woodworth BA, Xiong G, Ph D, Wolfe SG, Antunes MB, et al. Transient receptor potential vanilloid type 4 channel expression in chronic rhinosinusitis. Am J Rhinol. 2008;22(1):7–12.
Wu C. Distinct histopathology characteristics in empty nose syndrome. Laryngoscope. 2020;Epub ahead of print.
Alenmyr L, Herrmann A, Högestätt ED, Greiff L, Zygmunt PM. TRPV1 and TRPA1 stimulation induces MUC5B secretion in the human nasal airway in vivo. Clin Physiol Funct Imaging. 2011;31(6):435–44.
López-Requena A, Boonen B, Van Gerven L, Hellings PW, Alpizar YA, Talavera K. Roles of neuronal TRP channels in neuroimmune interactions. In: Neurobiology of TRP channels. 2017. p. Chapter 15.
Boonen B, Alpizar YA, Sanchez A, López-Requena A, Voets T, Talavera K. Differential effects of lipopolysaccharide on mouse sensory TRP channels. Cell Calcium. 2018;73:72–81.
Boonen B, Alpizar YA, Meseguer VM, Talavera K. TRP channels as sensors of bacterial endotoxins. Toxins (Basel). 2018;10(8).
Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun. 2014;5:3125.
Viana F. TRPA1 channels: molecular sentinels of cellular stress and tissue damage. J Physiol. 2016;594(15):4151–69.
Dux M, Rosta J, Messlinger K. TRP channels in the focus of trigeminal nociceptor sensitization contributing to primary headaches. Int J Mol Sci. 2020;21(1):342.
Quartu M, Serra MP, Boi M, Poddighe L, Picci C, Demontis R, et al. TRPV1 receptor in the human trigeminal ganglion and spinal nucleus: immunohistochemical localization and comparison with the neuropeptides CGRP and SP. J Anat. 2016;229(6):755–67.
Martinez JM, Eling TE. Activation of TRPA1 by volatile organic chemicals leading to sensory irritation. ALTEX. 2019;36(4):572–82.
Russell FA, King R, Smillie S, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2020;94(4):1099–142.
Kichko TI, Kobal G, Reeh XPW. Cigarette smoke has sensory effects through nicotinic and TRPA1 but not TRPV1 receptors on the isolated mouse trachea and larynx. Am J Physiol Lung Cell Mol Physiol. 2020;309(8):812–20.
Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D’Amours M, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci. 2009;106(22):9099–104.
Bautista DM, Pellegrino M, Tsunozaki M. TRPA1: a gatekeeper for inflammation. Annu Rev Physiol. 2013;75:181–200.
Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T, et al. Bimodal action of menthol on the transient receptor potential channel TRPA1. J Neurosci. 2007;27(37):9874–84.
Kichko TI, Neuhuber W, Kobal G, Reeh PW. The roles of TRPV1, TRPA1 and TRPM8 channels in chemical and thermal sensitivity of the mouse oral mucosa. Eur J Neurosci. 2018;47(3):201–10.
Urata K, Shinoda M, Ikutame D, Linuma T, Iwata K. Involvement of transient receptor potential vanilloid 2 in oral incisional pain. Oral Dis. 2018;24(6):1093–100.
Sato T, Sasahara N, Kanda N, Sasaki Y, Yamaguma Y, Kokubun S, et al. Distribution of CGRP and TRPV2 in human paranasal sinuses. Cells Tissues Organs. 2017;203(1):55–64.
Held K, Voets T, Vriens J. TRPM3 in temperature sensing and beyond. Temperature. 2015;2(2):201–13.
Yajima T, Sato T, Shimazaki K, Ichikawa H. Transient receptor potential melastatin-3 in the rat sensory ganglia of the trigeminal, glossopharyngeal and vagus nerves. J Chem Neuroanat 2019;96(September 2018):116–125.
Fujita M, Sato T, Yajima T, Masaki E, Ichikawa H. TRPC1, TRPC3, and TRPC4 in rat orofacial structures. Cells Tissues Organs. 2017;204(5–6):293–303.
Sanico AM, Koliatsos VE, Stanisz AM, Bienenstock J, Togias A. Neural hyperresponsiveness and nerve growth factor in allergic rhinitis. Int Arch Allergy Immunol. 1999;118:154–8.
Coffey CS, Mulligan RM, Schlosser RJ. Mucosal expression of nerve growth factor and brainderived neurotrophic factor in chronic rhinosinusitis. Am J Rhinol Allergy. 2009;23(6):571–4.
Yosipovitch G, Rosen JD, Hashimoto T. Itch: from mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol. 2018;142(5):1375–91.
Wallace H. Airway pathogenesis is linked to TRP channels. In: Neurobiology of TRP channels 2nd edition. 2017.
Turner P, Dear J, Scadding G, Foreman JC. Role of kinins in seasonal allergic rhinitis: icatibant, a bradykinin B2 receptor antagonist, abolishes the hyperresponsiveness and nasal eosinophilia induced by antigen. J Allergy Clin Immunol. 2001;107(1):105–13.
Dear J. Novel treatments for allergic rhinitis: an investigation into the role of bradykinin in the human nasal airway. Inflammopharmacology. 1996;4:225–39.
Fan HC, Zhang X, McNaughton PA. Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem. 2009;284(41):27884–91.
Lee K, Lee SH, Kim TH. The biology of prostaglandins and their role as a target for allergic airway disease therapy. Int J Mol Sci. 2020;21(5).
Li L, Guan K, Zhou Y, Wu J, Wang Y, Wang W. Prostaglandin E2 signal inhibits T regulatory cell differentiation during allergic rhinitis inflammation through EP4 receptor. World Allergy Organ J. 2019;12(12).
Taylor-Clark TE, Undem BJ, MacGlashan DW, Ghatta S, Carr MJ, McAlexander MA. Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1). Mol Pharmacol. 2008;73(2):274–81.
Materazzi S, Nassini R, Andrè E, Campi B, Amadesi S, Trevisani M, et al. Cox-dependent fatty acid metabolites cause pain through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A. 2008;105(33):12045–50.
Segboer CL, Holland CT, Reinartz SM, Terreehorst I, Gevorgyan A, Hellings PW, et al. Nasal hyper-reactivity is a common feature in both allergic and nonallergic rhinitis. Allergy. 2013;68(11):1427–34.
Tourangeau LM, Christiansen SC, Herschbach J, Brooks SM, Eddleston J, Zuraw B. Nasal mucosal TRPA1 and TRPV1 levels in human rhinitis. J Allergy Clin Immunol. 2011;127(2):AB52.
Greiff L, Svensson C, Andersson M, Persson CGA. Effects of topical capsaicin in seasonal allergic rhinitis. Thorax. 1995;50(3):225–9.
Kowalski ML, Dietrich-Miłobȩdzki A, Majkowska-Wojciechowska B, Jarzȩbska M. Nasal reactivity to capsaicin in patients with seasonal allergic rhinitis during and after the pollen season. Allergy. 1999;54(8):804–10.
Alenmyr L, Högestätt ED, Zygmunt PM, Greiff L. TRPV1-mediated itch in seasonal allergic rhinitis. Allergy. 2009;64(5):807–10.
Gerth Van Wijk R, Terreehorst IT, Mulder PGH, Garrelds IM, Blom HM, Popering S. Intranasal capsaicin is lacking therapeutic effect in perennial allergic rhinitis to house dust mite. A placebo-controlled study. Clin Exp Allergy. 2000;30(12):1792–8.
Kortekaas Krohn I, Callebaut I, Alpizar YA, Steelant B, Van Gerven L, Skov PS, et al. MP29-02 reduces nasal hyperreactivity and nasal mediators in patients with house dust mite-allergic rhinitis. Allergy Eur J Allergy Clin Immunol. 2018;73(5):1084–93.
Alenmyr L, Greiff L, Andersson M, Sterner O, Zygmunt PM, Högestätt ED. Effect of mucosal TRPV1 inhibition in allergic rhinitis. Basic Clin Pharmacol Toxicol. 2012;110(3):264–8.
Bareille P, Murdoch RD, Denyer J, Bentley J, Smart K, Yarnall K, et al. The effects of a TRPV1 antagonist, SB- 705498, in the treatment of seasonal allergic rhinitis. Int J Clin Pharmacol Ther. 2013;51(7):576–84.
• Van Gerven L, Steelant B, Cools L, Callebaut I, Backaert W, de Hoon J, et al. Low-dose capsaicin (0.01 mM) nasal spray is equally effective as the current standard treatment for idiopathic rhinitis: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2020;3. Rationale behind capsaicin treatment for nasal hyperreactivity and protocol for capsaicin therapy in daily practice.
Everaerts W, Gees M, Alpizar YA, Farre R, Leten C, Apetrei A, et al. The capsaicin receptor TRPV1 is a crucial mediator of the noxious effects of mustard oil. Curr Biol. 2011;21(4):316–21.
Gees M, Alpizar YA, Boonen B, Sanchez A, Everaerts W, Segal A, et al. Mechanisms of transient receptor potential vanilloid 1 activation and sensitization by allyl isothiocyanate. Mol Pharmacol. 2013;84(3):325–34.
Alpizar YA, Boonen B, Gees M, Sanchez A, Nilius B, Voets T, et al. Allyl isothiocyanate sensitizes TRPV1 to heat stimulation. Pflugers Arch Eur J Physiol. 2014;466(3):507–15.
Tóth E, Tornóczky T, Kneif J, Perkecz A, Piski Z, Kemény Á, et al. Upregulation of extraneuronal TRPV1 expression in chronic rhinosinusitis with nasal polyps. Rhinology. 2018;56(3):245–54.
Kim J-H, Lee J-S, Jang Y-S, Park JY, Hwang Y Il, Park S, et al. The expression of TRPV1 and innate Th2-cytokines in patients with chronic rhinosinusitis and asthma. J Allergy Clin Immunol. 2018;141(2):AB115.
Xu R, Li Q, Zhou J, Zhou X, Perelman JM, Victor P. The degradation of airway tight junction protein under acidic conditions is probably mediated by transient receptor potential vanilloid 1 receptor. Biosci Rep. 2013;33(5):847–56.
Talavera K, Startek JB, Alvarez-Collazo J, Boonen B, Alpizar YA, Sanchez A, et al. Mammalian transient receptor potential TRPA1 channels: from structure to disease. Physiol Rev. 2020;100(2):725–803.
Maher SA, Dubuis ED, Belvisi MG. G-protein coupled receptors regulating cough. Curr Opin Pharmacol. 2011;11(3):248–53.
Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Högestätt ED, et al. Pungent products from garlic activate the sensory ion channel TRPA1. Proc Natl Acad Sci U S A. 2005;102(34):12248–52.
Van Gerven L, Boeckxstaens G, Jorissen M, Fokkens W, Hellings PW. Short-time cold dry air exposure: a useful diagnostic tool for nasal hyperresponsiveness. Laryngoscope. 2012;122(12):2615–20.
Miyake T, Nakamura S, Zhao M, So K, Inoue K, Numata T, et al. Cold sensitivity of TRPA1 is unveiled by the prolyl hydroxylation blockade-induced sensitization to ROS. Nat Commun. 2016;7(12840).
Lawhorn BG, Brnardic EJ, Behm DJ. Recent advances in TRPV4 agonists and antagonists. Bioorganic Med Chem Lett. 2020;30(8).
Grace MS, Bonvini SJ, Belvisi MG, McIntyre P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol Ther. 2017;177:9–22.
Rosenbaum T, Benítez-Angeles M, Sánchez-Hernández R, Morales-Lázaro SL, Hiriart M, Morales-Buenrostro LE, et al. TRPV4: a physio and pathophysiologically significant ion channel. Int J Mol Sci. 2020;21(11):3837.
Liu SC, Lu HH, Cheng LH, Chu YH, Lee FP, Wu CC, et al. Identification of the cold receptor TRPM8 in the nasal mucosa. Am J Rhinol Allergy. 2015;29(4):e112–6.
Keh SM, Facer P, Yehia A, Sandhu G, Saleh HA, Anand P. The menthol and cold sensation receptor TRPM8 in normal human nasal mucosa and rhinitis. Rhinology. 2011;49(4):11.
Alonso-Carbajo L, Alpizar YA, Startek JB, López-López JR, Pérez-García MT, Talavera K. Activation of the cation channel TRPM3 in perivascular nerves induces vasodilation of resistance arteries. J Mol Cell Cardiol. 2019;129:219–30.
Tang R, Li ZP, Li MX, Li DW, Ye HB, Su KM, et al. Pro-inflammatory role of transient receptor potential canonical channel 6 in the pathogenesis of chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2018;8(11):1334–41.
Fu Z, Gu L, Li N, Ma Z, Ling M, Wang Y. Upregulated TRPC5 plays an important role in development of nasal polyps by activating eosinophilic inflammation and NF-κB signaling pathways. Int J Clin Exp Pathol. 2018;11(4):1935–45.
Funding
The laboratory of Peter Hellings is supported by a KU Leuven grant (C14/18/086).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Rhinosinusitis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Backaert, W., Steelant, B., Hellings, P.W. et al. A TRiP Through the Roles of Transient Receptor Potential Cation Channels in Type 2 Upper Airway Inflammation. Curr Allergy Asthma Rep 21, 20 (2021). https://doi.org/10.1007/s11882-020-00981-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s11882-020-00981-x