
Opinion statement
Autophagy is a physiological process that occurs in normal tissues. Under external environmental pressure or internal environmental changes, cells can digest part of their contents through autophagy in order to reduce metabolic pressure or remove damaged organelles. In cancer, autophagy plays a paradoxical role, acting as a tumor suppressor—by removing damaged organelles and inhibiting inflammation or by promoting genome stability and the tumor-adaptive responses—as a pro-survival mechanism to protect cells from stress. In this article, we review the autophagy-dependent mechanisms driving childhood central nervous system tumor cell death, malignancy invasion, chemosensitivity, and radiosensitivity. Autophagy inhibitors and inducers have been developed, and encouraging results have been achieved in autophagy modulation, suggesting that these might be potential therapeutic agents for the treatment of pediatric central nervous system (CNS) tumors.
Similar content being viewed by others


Introduction
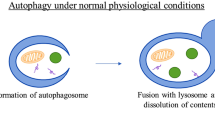
Autophagy is a catabolic process that captures and degrades damaged proteins and organelles in lysosomes [1, 2]. It effectively degrades normal cell metabolites and helps to maintain the health of the body, and in some cases autophagy can selectively remove cellular components such as damaged or excess peroxisomes, endoplasmic reticulum, mitochondria, or DNA [3••] thereby reducing the accumulation of abnormal proteins and organelles and maintaining cell homeostasis. The role of autophagy in promoting survival and maintaining cell homeostasis has been widely confirmed at the cell and organ level. For example, selectively knocking out the Atg5 or Atg7 genes in the brain leads to the accumulation of polyubiquitinated proteins and neuronal degeneration in mice. The survival and proliferation of T cells depends on Atg5 [4], and Atg5-deficient mice cannot survive during the neonatal period and their tissues show the presence of amino acid exhaustion and insufficient metabolism [5]. Finally, in the absence of growth factors, the autophagy of hematopoietic cells is significantly enhanced in order to maintain the ATP supply and cell survival.
Autophagy is a physiological process that occurs in normal tissues. When faced with external environmental pressures (e.g., amino acid deficiency, insufficient glucose supply, or reduced oxygen supply) or internal environmental changes (e.g., protein, DNA, or mitochondrial damage or microbial infection), cells can digest part of their contents through autophagy in order to resist metabolic pressure or to remove damaged organelles [6]. Basal levels of autophagy play an important role in maintaining homeostasis in normal tissues, while nutrient deficiency, hypoxia, DNA damage, and cytotoxicity can all induce increased levels of autophagy. The induction of autophagy can promote cell survival by adjusting the dynamic balance inside and outside the cell [7]; however, excessive autophagy can seriously affect embryonic differentiation and induce cell death. Therefore, changes in autophagy are closely related to various clinical diseases such as cancer, neurodegeneration, heart disease, liver and metabolic disorders, infectious diseases, and autoimmune diseases [8••, 9, 10]. In the past decade, autophagy has received more and more attention as a new target in the treatment of various diseases.
Autophagy and tumors
Malignant tumors are an important cause of childhood death worldwide, and their incidence has increased in recent years. Although current anti-tumor therapies have led to a positive prognosis for many patients, the efficacy of these therapies is still limited for some tumors. Autophagy occurs frequently during tumorigenesis, and at different stages of tumor development autophagy may have opposite effects, such as being a tumor suppressor or a tumor-initiating factor. In general, autophagy protects cancer cells during chemotherapy, and this can easily lead to tumor resistance and the development of refractory cancers [11]. Many external stimuli can affect tumor autophagy, such as hypoxia, acidification of the tumor microenvironment, nutritional deficiencies, drug treatment, or infection [12]. In addition, tumor suppressors or oncogenes can regulate the autophagy pathway in cancer cells. For example, p53 status can change the role of autophagy in tumor progression [13, 14].
Tumor-suppressing roles for autophagy
Autophagy was originally considered to be a tumor suppressor mechanism. This concept originated from early reports that the crucial autophagy gene BECN1 is lost in 40–75% of human prostate, breast, and ovarian cancers [15, 16]. In genetic knockout mouse models of hereditary breast cancer, the deletion of BECN1 promotes the activation of p53 and reduces the occurrence of tumors. However, the deletion of BECN1 in human cancers and the deletion of the breast cancer 1 (BRCA1) gene cannot be disassociated, which indicates that BECN1 is not a tumor suppressor in most human cancers. Thus, BECN1 only exerts a tumor suppressor effect in genetic animal models of cancer [15].
The basal level of autophagy inhibits the occurrence of tumors by controlling the degradation of damaged components and proteins in cells [17]. Autophagy in genetic knockout mouse models can inhibit the accumulation of reactive oxygen species (ROS), DNA damage, tissue damage, inflammation, genome instability, and other tumor-initiating factors and thus can suppress tumors in their early stages [18]. Damage to mitochondria can lead to excessive ROS production, thereby promoting carcinogenesis, and autophagy prevents the occurrence of tumors by removing these malfunctioning mitochondria [19]. In mice lacking autophagy due to knockout of both Atg5 and Atg7, oxidative stress and mitochondrial damage can induce hepatocytes to form liver tumors [20]. In genetic knockout mouse models of lung cancer, breast cancer, pancreatic cancer, and melanoma, deletion of the autophagy gene Ras or Braf inhibits the growth of benign tumors but accelerates the growth of malignant tumors. When exposed to chemical carcinogens, Atg4-deficient mice are more susceptible to fibrosarcoma [8••, 21]. Other studies have shown that the loss of autophagy-related genes such as Atg3, Atg5, and Atg9 is also related to tumorigenesis [20]. Taken together, these results suggest that autophagy is an important mechanism for inhibiting tumor growth and that impaired autophagy can lead to tumor formation (Fig. 1, left panel).

Tumor-suppressing and tumor-promoting roles for autophagy in cancer. The left panel shows the proposed mechanisms through which autophagy may suppress tumors by regulating oncogenic proteins, genomic stability, cell proliferation, cell death mechanisms, stress-related responses, and immune-response mechanisms. The right panel shows the proposed mechanisms for the tumor-promoting effect of autophagy by providing nutrients and energy to cancer cells, adaptation to oxidative stress and DNA damage, angiogenesis, metastasis and invasion during tumorigenesis, the unfolded protein response in cancer cells, tumor growth, and resistance to chemotherapy drugs in cancer cells.
Tumor-promoting roles for autophagy
While basal levels of autophagy are low in normal cells and tissues, many cancer cell lines show high levels of autophagy [22•]. Autophagy promotes tumor growth and survival and the development of malignant tumors by maintaining the basic metabolic functions of tumor cells [23]. In addition, autophagy meets the metabolic needs of tumor cells by increasing stress tolerance and providing nutrients, and it maintains cell survival even under adverse conditions such as starvation or hypoxia, which are extremely common during tumor growth [18, 23]. Studies have confirmed that autophagy is upregulated in hypoxic tumor areas in the tumor microenvironment, thus inhibiting tumor-induced inflammation and promoting tumor cell survival [24]. Therefore, if tumor-promoting pathways are activated due to stress in tumor cells or to stress in the tumor microenvironment, this will increase the demand for autophagy, thereby promoting the growth and survival of tumors [25].
Studies have found that RAS mutations in tumor cells increase the level of autophagy, which can enhance tumor growth, survival, and deterioration, and such mutations are associated with the development of some cancers, including lung, colon, and pancreatic cancer [26,27,28]. Among the genetic knockout mouse models of lung cancer, pancreatic ductal adenocarcinoma, prostate cancer, and melanoma, tumors that were missing Atg5 or Atg7 grew slowly [29,30,31]. The deletion of BECN1 in breast cancer cells and the deletion of ATG13 or ULK1 in glioblastoma cells led to similar results [32, 33]. Thus, inhibition of autophagy can induce tumor cell death.
Loss of Atg5 or Atg7 in mice can cause chronic liver damage, inflammation, and benign liver tumors, but these fail to develop into cancer [18]. As these tumors remain benign, this indicates that even though depletion of autophagy can increase tumor initiation in the livers of mice, autophagy is necessary for tumors to develop to the malignant stage [8••]. Specifically knocking out autophagy genes in mice can promote the formation of tissue damage and inflammation-related benign lesions, but many aggressive cancer cells require autophagy to grow and survive [34, 35]. The epithelial mesenchymal transition (EMT) plays an important role in tumor metastasis, and autophagy and EMT are interrelated [36, 37]. During tumor metastasis, cancer cells activated by EMT show high levels of autophagy and can survive in a variety of stressful conditions [38, 39]. Thus, autophagy can promote tumor development by promoting cancer cell proliferation and tumor growth (Fig. 1, right panel).
The role of autophagy in childhood CNS tumors
CNS tumors are common solid tumors in children, with an average annual incidence of 1.7–4.1 per 100,000, which has increased slightly in recent decades and is second only to leukemia in terms of childhood tumors [40]. In recent years, despite great progress in early diagnosis, surgical procedures, and treatment strategies, the overall prognosis of CNS tumors is still very poor, with a 5-year survival rate of only 33% [41]. The histological analysis of childhood primary brain tumors and other CNS tumors in children between the ages of 0 and 14 years in the Central Brain Tumor Registry of the United States (CBTRUS) showed that the most common childhood CNS tumors are gliomas, medulloblastomas, atypical teratoid/rhabdoid tumors (ATRTs), craniopharyngiomas, and ependymomas. These are all malignant tumors, accounting for 80% of children’s CNS tumors [40, 42], and among all CNS tumors in children more than 90% are malignant [43].
Although childhood hematological tumors such as leukemia are still the main research focus for clinicians, CNS tumors have received more and more attention in recent years. The typical treatments of childhood CNS tumors, such as surgery, chemotherapy, and radiotherapy, have greatly improved the survival rate of children with CNS tumors. However, there are still some patients who respond poorly to treatment, and clinicians still need to find new therapeutic targets. Autophagy as a potential therapeutic target has been studied in a variety of cancers, which may provide a new therapeutic strategy for childhood CNS tumor patients. In this article, we focus on the most recent advances in the context of autophagy-dependent mechanisms driving childhood CNS tumor cell death, invasiveness, chemosensitivity, and radiosensitivity together with autophagy modulator treatment strategies that can be used to overcome autophagy-mediated drug resistance.
Autophagy and cell death in childhood CNS tumors
Autophagy and cell death are two crucial cellular processes with complex protein networks, and there is a certain overlap in the regulatory mechanisms between them [44,45,46,47]. Studies have suggested that autophagy may serve as an alternative pathway to inducing cell death in many tumor cells with defects in apoptosis. For example, in BAX and BAK-deficient cancer cells endoplasmic reticulum stress-responsive apoptosis is prevented, but sustained autophagy can cause oxidative damage-induced cell death [44]. The autophagy and apoptosis signaling pathways are both separate and interconnected. Initially, autophagy and apoptosis were thought to differ as modes of cellular degradation in terms of morphology, biochemical indicators, molecules, and mechanisms. Later evidence showed that the two pathways can antagonize or promote each other under certain situations.
Autophagy was shown to be required for glioblastoma development in mice, and in light of the high resistance of malignant gliomas to apoptosis the induction of autophagic cell death by autophagy stimulators is an alternative method for triggering cell death in glioblastomas [48]. Temozolomide can induce apoptosis through the selective inhibition of autophagy, in which autophagic vehicles accumulate because their fusion with lysosomes is blocked. Modulation of the autophagic action of temozolomide with autophagy inhibitors can result in opposite outcomes depending on the step in the autophagic pathway that is targeted [49]. Su et al. identified a novel potential RAB13 inhibitor, which was confirmed to negatively regulate autophagy and induce cell death in low-grade glioma cells [50].
In other common type of childhood CNS tumors, the significant induction of autophagy produced by pimozide, a neuroleptic drug used for the treatment of schizophrenia and chronic psychosis, can promote medulloblastoma cell apoptosis by inhibiting the expression of the anti-apoptotic markers c-Myc, Mcl-1, and Bcl-2 [51]. In another in vitro study, plant-derived Δ9-tetrahydrocannabinol and cannabidiol induced cell cycle arrest in medulloblastoma and ependymoma cells in part through the production of ROS and the induction of autophagy and apoptosis [52].
Overall, these studies suggest a role for autophagy in regulating apoptosis in childhood CNS tumors. However, due to the complex network between autophagy and apoptosis, the effects of tumor–stroma interactions, and differences in the tumor microenvironment, more appropriate mouse models are required in order to clarify the function of autophagy in the apoptosis of CNS tumors in children.
Autophagy and malignancy invasion in childhood CNS tumors
Childhood CNS tumors can acquire invasive properties by undergoing EMT, and this allows them to infiltrate into the surrounding normal brain tissue thus preventing complete surgical resection of the tumor. The autophagic process is closely related to tumorigenesis and the development of malignancies, and autophagy may have impacts on the invasiveness of brain tumors in children [53, 54].
Decreasing N-cadherin expression in glioblastoma cells has been shown to impair their focal adhesion and enhance their migratory capacity [55], and an in vitro study of glioblastoma autophagy has been shown to facilitate the degradation of SNAIL family proteins leading to upregulation of the N-cadherin level, which suggests that autophagy may suppress the invasive properties of glioblastomas [56, 57]. In addition, autophagy has also been reported to be involved in the regulation of the Wnt signaling pathway and the RKT Met signaling pathway, which are involved in the regulation of glioblastoma cell invasion through their effects on N-cadherin and vascular endothelial growth factor [58,59,60]. These findings demonstrated that autophagy can modulate different signaling pathways in glioblastoma cell lines, and it would be interesting to determine how this regulatory network can influence the invasion of glioblastoma cells. To investigate the effect of autophagy on the invasion of medulloblastoma, a recent study used shRNA-mediated knockdown of ATG5 in medulloblastoma cell lines belonging to the SHH group 3 and group 4 subtypes. Their findings showed that autophagy inhibition did not result in a significant difference in the proliferation or anchorage-independent growth of the medulloblastoma cells; however, autophagy inhibition led to a substantial reduction in the invasive potential of all three medulloblastoma cell lines [61]. In another study, the role of the pro-autophagy factor AMBRA1 in regulating medulloblastoma was identified showing that AMBRA1 expression depends on c-MYC levels and is correlated with poor prognosis in group 3 patients. Knockdown of AMBRA1 reduced the stemness, growth, and invasiveness of group 3 medulloblastoma stem cells [62]. Thus, it appears that regulation of autophagy profoundly affects the invasive potential and growth of childhood CNS tumor cells, which suggests a therapeutic potential for autophagy modulators in the treatment of pediatric brain tumors.
Autophagy and chemosensitivity in childhood CNS tumors
Chemotherapy is still the main treatment for pediatric brain tumors; however, the prognosis of chemotherapy treatment in some groups of high-risk patients remains dismal. Intrinsic or acquired chemoresistance to chemotherapy drugs is a major clinical obstacle to the treatment of childhood CNS tumor patients. Therefore, a better understanding of the molecular mechanisms underlying chemoresistance to chemotherapy drugs may lead to improved clinical outcomes in pediatric brain tumor patients.
Recent studies have shown that modulation of autophagy in response to chemotherapy drug treatment, such as temozolomide, may hold great promise for circumventing chemotherapeutic resistance and improving anticancer efficacy in brain tumor patients [63, 64]. Li and colleagues demonstrated that the sensitivity of glioblastoma cells to temozolomide was increased by miR-519a, which might be mediated through autophagy, and that miR-519a overexpression could induce autophagy by inhibiting the STAT3/Bcl-2 pathway [65]. In another study, the authors used siRNA to knock down the autophagy-related genes ATG12 and ATG7 and then pharmacologically induced or inhibited these genes using rapamycin or chloroquine, respectively, to test the effect of autophagy on chemosensitivity in pediatric ATRT cell lines (BT-16 and BT-12). They found that silencing ATG12 and ATG7 or exposing the cells to the autophagy inhibitor chloroquine could inhibit this increase in autophagy; however, the effect of autophagy on killing tumor cells was minimal [66]. A recent study reported an oncogenic role for the nucleoporin TPR (translocated promoter region, a nuclear basket protein) in regulating heat shock transcription factor 1 (HSF1) mRNA trafficking, maintaining MTORC1 activity to phosphorylate ULK1, and preventing macroautophagy/autophagy induction in ependymomas. The authors found that high expression of TPR was associated with increased HSF1 and HSPA/HSP70 expression in ependymoma patients and showed that MTOR inhibition by rapamycin therapeutically suppressed TPR expression and reduced tumor size in an ependymoma mouse xenograft model [67]. These studies included both in vivo and in vitro experiments, and the findings showed that chemosensitivity in childhood brain tumors could be regulated by modulating autophagy levels, and this may have clinical relevance in the future planning of therapeutic regimens for pediatric brain tumors. However, autophagy is a dynamic process with multiple steps involved in producing autophagosomes, fusing with lysosomes, and completing the degradation of intra-vesicular contents, and it is feasible that blocking autophagosome formation has different effects on tumor cell survival than blocking autophagic flux. More research is needed to elucidate the mechanisms by which autophagy modulates the different chemotherapeutic agents, but at least for now autophagy as a potential therapeutic target provides new strategies for the treatment of childhood CNS tumors.
Autophagy and radiosensitivity in childhood CNS tumors
Due to the local growth patterns of childhood CNS tumors, complete surgical removal is difficult in many patients and postoperative radiotherapy is necessary. Recently, autophagy has been reported to be involved in the regulation of radiosensitivity in childhood CNS tumors in both in vivo and in vitro studies [68,69,70], which suggests a new treatment strategy for pediatric brain tumor patients who are sensitive to radiation therapy. In a recent study, the authors assessed FOXG1 expression in glioma tissues and glioma-adjacent tissues, and they found that the FOXG1 expression level was up-regulated in glioma cells following exposure to irradiation and that FOXG1 reduced the radiosensitivity of glioma cells by promoting autophagy [68]. Lee et al. assessed the therapeutic effects of combining disulfiram with radiation treatment in ATRT cells (SNU.ATRT-5 and SNU.ATRT-6) and showed that disulfiram enhanced the radiosensitivity of ATRT cells with a reduction in the survival fraction and increased DNA double-strand breaks, apoptosis, autophagy, and cell cycle arrest in irradiated ATRT cells [69]. These studies suggest that autophagy might be a novel modulator for those childhood CNS tumor patients who receive radiotherapy; however, the outcome of autophagy observed in brain tumors after radiotherapy is not straightforward. Although an association between autophagy and radiosensitization was demonstrated, the precise role of autophagy in relation to brain tumor cell death is hard to define and more comprehensive analyses are needed.
Autophagy modulators in childhood CNS tumors
Evidence for the effects of pharmaceutical modulators of autophagy in pediatric brain tumors is limited; however, the antitumor effect of autophagy remains an exciting potential treatment strategy. The BRAF (V600E) mutation is important in childhood CNS tumors [71], and a study showed that the autophagy inhibitor chloroquine reduced tumor viability in glioma cells with the BRAF (V600E) mutation [72]. The authors also demonstrated that chloroquine could improve vemurafenib sensitivity in children with ganglioglioma, indicating that pediatric CNS tumors with BRAF (V600E) are autophagy-dependent and should be targeted with autophagy inhibition in combination with other therapeutic strategies. Another autophagy modulator, salinomycin, can induce ROS in abortive autophagy, and this leads to regulated necrosis in glioblastoma cells [73]. For medulloblastoma, MirR-30a inhibited autophagy by reducing beclin 1/ATG5 expression and was linked to increased cell death in a medulloblastoma cell line [74]. Until now, there have only been a few clinical trials for autophagy modulators in treatment of childhood CNS tumors [75] (Table 1). Hydroxychloroquine combined with Dabrafenib (NCT04201457), everolimus (NCT00187174), everolimus combined with lenvatinib (NCT03245151), and Temsirolimus combined with valproic acid (NCT01204450) were used in clinical trials as modulators of induced autophagy in the treatment of childhood CNS tumors [76,77,78,79]. These clinical trials provide further evidence for autophagy modulators as potential therapeutic agents for the treatment of childhood CNS tumors.
Autophagy and CNS tumor treatment-related brain injury
At present, the treatment of pediatric CNS tumors is still mainly based on surgery, radiotherapy, and chemotherapy. In particular, the related complications after multiple radiotherapy and/or chemotherapy sessions are still crucial issues that cannot be ignored in the clinic.
Neurotoxic brain injury caused by some anti-tumor chemotherapy drugs occurs widely in the clinic, and this reduces the treatment effect and quality of life of patients, especially for growing and developing children with CNS tumors [80]. Severe neurotoxic responses often result in patients facing the dilemma of reducing the dose of chemotherapeutic drugs, while at the same time such responses negatively impact on the patient's mental and physical state and their quality of life. Autophagy can regulate the extent of neurotoxic brain injury caused by anti-tumor drugs. For example, the peripheral nervous system damage caused by bortezomib can also enhance the level of autophagy [81]. Bortezomib is a proteasome inhibitor that prevents the degradation of misfolded proteins in the nervous system, resulting in severe neurotoxicity. Bortezomib can also activate the transcription factor ATF4 and up-regulate the expression of LC3-II, thereby activating autophagy [82]. By enhancing autophagy, proteasome inhibition can degrade protein aggregates and interfere with the neurotoxicity caused by bortezomib [83].
Radiotherapy is one of the most effective tools in the treatment of pediatric CNS tumors. However, damage to normal brain tissue surrounding the tumor constitutes a major problem and is associated with adverse side effects, particularly in pediatric patients. Autophagy is essential for survival, differentiation, development, and homeostasis [46, 84, 85], but inappropriate activation of autophagy is directly involved in triggering the initiation of apoptotic or necrotic cell death [86]. In an in vitro study, neural stem cells were shown to be extremely sensitive to irradiation [87]. Atg7 knockdown significantly decreased autophagy, thus increasing apoptosis levels in irradiated neural stem cells, suggesting that autophagy protects NSCs from radiation-induced apoptosis. This indicated that downregulating autophagy by selective Atg7 knockdown in NSCs enhances radiation-induced neural stem cell damage, thus suggesting an important protective role for autophagy in maintaining neurogenesis. However, in a recent in vivo study using 10-day-old selective Atg7 knockout mice subjected to a single 6 Gy dose of whole-brain irradiation, cell death and proliferation, microglia activation, and inflammation were reduced compared to wild type mice in the acute phase after irradiation. Selective neural deletion of the Atg7 gene reduced irradiation-induced cerebellar white matter injury in the juvenile mouse brain by ameliorating oligodendrocyte progenitor cell loss in the subacute phase after irradiation [88, 89]. Together, the conflicting roles played by autophagy in different experimental conditions led to different protective effects of autophagy on neural stem cells in the above mentioned in vivo and in vitro studies. But at least, these results suggest that autophagy might be a potential target for brain injury after radiotherapy in pediatric CNS tumor patients.
Conclusion
Numerous autophagy inhibitors and inducers have been developed, and encouraging results have been achieved in autophagy modulation. Preclinical trials of autophagy inhibitors and inducers combined with chemotherapeutics or radiotherapy may improve their efficacy and their therapeutic effects in cancer patients; however, clinical trials focusing on autophagy control are limited to only a few autophagy promoters and inhibitors. Therefore, further research is needed to determine their anti-tumor efficacy, including autophagy modulators and chemotherapeutic drugs that are used for pediatric CNS tumor patients. In addition, more research must be conducted in order to develop specific therapeutic agents for the treatment of pediatric CNS tumors.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Galluzzi L, et al. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–99.
Dikic I, et al. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–64.
•• Mizushima N, et al. Autophagy in human diseases. N Engl J Med. 2020;383(16): 1564-76. This reference reviewed and discussed large preclinical data linking autophagy dysfunction to the pathogenesis of major human disorders including cancer as well as other common diseases.
Qiao CM, et al. Sodium butyrate causes alpha-synuclein degradation by an Atg5-dependent and PI3K/Akt/mTOR-related autophagy pathway. Exp Cell Res. 2020;387(1):111772.
Feng X, et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy. 2021;17(3):723–42.
Parzych KR, et al. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–73.
Kim KH, et al. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–37.
•• Onorati AV, et al. Targeting autophagy in cancer. Cancer 2018;124(16): 3307-18. This reference summarized fundamental advances in the biology of autophagy, approaches to targeting autophagy, the preclinical rationale and clinical experience with hydroxychloroquine in cancer clinical trials, the potential role of autophagy in tumor immunity, and recent developments in next-generation autophagy inhibitors that have clinical potential.
Menzies FM, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–34.
Dong Y, et al. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol. 2019;136:27–41.
Jiang GM, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18(1):17.
Butera G, et al. Regulation of autophagy by nuclear GAPDH and its aggregates in cancer and neurodegenerative disorders. Int J Mol Sci. 2019;20(9):2062.
Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300.
Yang A, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4(8):905–13.
White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–6.
Joffre C, et al. [The yin and the yang of autophagy in cancer cells], Med Sci (Paris). 2017;33(3):328–34.
White E, et al. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–46.
Poillet-Perez L, et al. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019;33(11-12):610–9.
Gao L, et al. Targeting ROS-mediated crosstalk between autophagy and apoptosis in cancer. Adv Exp Med Biol. 2020;1260:1–12.
Yun CW, et al. The roles of autophagy in cancer. Int J Mol Sci. 2018;19(11):3466.
Levy JMM, et al. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
• Udristioiu A, et al. Autophagy dysfunctions associated with cancer cells and their therapeutic implications. Biomed Pharmacother. 2019;115:108892 This reference reviewed recent insights of the molecular mechanism of autophagy and the potential roles of autophagy in cell death, cancer development, overview of the most recent therapeutic strategies involving autophagy modulators in cancer prevention and therapeutic opportunities.
Kimmelman AC, et al. Autophagy and tumor metabolism. Cell Metab. 2017;25(5):1037–43.
Mowers EE, et al. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018;285(10):1751–66.
Camuzard O, et al. Autophagy in the crosstalk between tumor and microenvironment. Cancer Lett. 2020;490:143–53.
Wang Y, et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy. 2019;15(5):886–99.
Bryant KL, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–40.
Chen P, et al. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J Exp Clin Cancer Res. 2019;38(1):254.
Zhang P, et al. ATG7-dependent and independent autophagy determine the type of treatment in lung cancer. Pharmacol Res. 2021;163:105324.
Santanam U, et al. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016;30(4):399–407.
Wang ZC, et al. MicroRNA-137 inhibits autophagy and chemosensitizes pancreatic cancer cells by targeting ATG5. Int J Biochem Cell Biol. 2019;111:63–71.
Wijshake T, et al. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc Natl Acad Sci U S A. 2021;118(5):e2020478118.
Gammoh N, et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy. 2016;12(9):1431–9.
Kuma A, et al. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13(10):1619–28.
Amaravadi RK, et al. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9(9):1167–81.
Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412.
Yeung KT, et al. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017;11(1):28–39.
Gao T, et al. Long non-coding RNA 91H regulates IGF2 expression by interacting with IGF2BP2 and promotes tumorigenesis in colorectal cancer. Artif Cells Nanomed Biotechnol. 2020;48(1):664–71.
Pastushenko I, et al. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–26.
Louis DN, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–20.
Fruhwald MC, et al. Tumors of the central nervous system in children and adolescents. Dtsch Arztebl Int. 2011;108(22):390–7.
Udaka YT, et al. Pediatric Brain Tumors. Neurol Clin. 2018;36(3):533–56.
Collaborators GBaOCC Global, regional, and national burden of brain and other CNS cancer, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(4): 376-93.
Mukhopadhyay S, et al. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19(4):555–66.
D'arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–92.
Yan X, et al. Autophagy-cell survival and death. Adv Exp Med Biol. 2019;1206:667–96.
Cheng Y, et al. Autophagy and tumor cell death. Adv Exp Med Biol. 2020;1207:339–49.
Lefranc F, et al. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12(12):1395–403.
Pawlowska E, et al. An interplay between senescence, apoptosis and autophagy in glioblastoma multiforme-role in pathogenesis and therapeutic perspective. Int J Mol Sci. 2018;19(3):889.
Su W, et al. Identification of autophagic target RAB13 with small-molecule inhibitor in low-grade glioma via integrated multi-omics approaches coupled with virtual screening of traditional Chinese medicine databases. Cell Prolif. 2021:e13135.
Ranjan A, et al. Pimozide suppresses the growth of brain tumors by targeting STAT3-mediated autophagy. Cells. 2020;9(9):2141.
Andradas C, et al. Assessment of cannabidiol and Delta9-Tetrahydrocannabiol in mouse models of medulloblastoma and ependymoma. Cancers (Basel). 2021;13(2):330.
Dongre A, et al. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
Carvajal L, et al. Autophagy process in trophoblast cells invasion and differentiation: similitude and differences with cancer cells. Front Oncol. 2021;11:637594.
Camand E, et al. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J Cell Sci. 2012;125(Pt 4):844–57.
Catalano M, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–25.
Asano K, et al. Correlation of N-cadherin expression in high grade gliomas with tissue invasion. J Neurooncol. 2004;70(1):3–15.
Colella B, et al. Autophagy induction impairs Wnt/beta-catenin signalling through beta-catenin relocalisation in glioblastoma cells. Cell Signal. 2019;53:357–64.
Barrow-Mcgee R, et al. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat Commun. 2016;7:11942.
Piao Y, et al. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro Oncol. 2016;18(9):1230–41.
Paul R, et al. Autophagy inhibition impairs the invasion potential of medulloblastoma cells. Mol Biol Rep. 2020;47(7):5673–80.
Nazio F, et al. Targeting cancer stem cells in medulloblastoma by inhibiting AMBRA1 dual function in autophagy and STAT3 signalling. Acta Neuropathol. 2021;142:537–64.
Daido S, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64(12):4286–93.
Xu X, et al. Association between SOX9 and CA9 in glioma, and its effects on chemosensitivity to TMZ. Int J Oncol. 2018;53(1):189–202.
Li H, et al. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol. 2018;11(1):70.
Levy JM, et al. Modulation of pediatric brain tumor autophagy and chemosensitivity. J Neurooncol. 2012;106(2):281–90.
Dewi FRP, et al. Nucleoporin TPR (translocated promoter region, nuclear basket protein) upregulation alters MTOR-HSF1 trails and suppresses autophagy induction in ependymoma. Autophagy. 2021;17(4):1001–12.
Xiao N, et al. FOXG1 mediates the radiosensitivity of glioma cells through regulation of autophagy. Int J Radiat Biol. 2021;97(2):139–48.
Lee YE, et al. Repositioning disulfiram as a radiosensitizer against atypical teratoid/rhabdoid tumor. Neuro Oncol. 2017;19(8):1079–87.
Marampon F, et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017;397:1–11.
Packer RJ, et al. Molecular-targeted therapy for childhood brain tumors: a moving target. J Child Neurol. 2020;35(12):791–8.
Levy JM, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–80.
Xipell E, et al. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget. 2016;7(21):30626–41.
Singh SV, et al. Restoration of miR-30a expression inhibits growth, tumorigenicity of medulloblastoma cells accompanied by autophagy inhibition. Biochem Biophys Res Commun. 2017;491(4):946–52.
Gatto F, et al. Recent advances in understanding the role of autophagy in paediatric brain tumours. Diagnostics (Basel). 2021;11(3):481.
Hoffman L. A trial of dabrafenib, trametinib and hydroxychloroquine for patients with recurrent LGG or HGG with a BRAF aberration. 2019. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04201457?term=NCT04201457&draw=2&rank=1. Accessed 17 Dec 2019.
Furman WL. Everolimus for treating pediatric patients with recurrent or refractory tumors. 2005. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT00187174?term=NCT00187174&draw=2&rank=1. Accessed 16 Sept 2005.
Inc. E, et al. Study of lenvatinib in combination with everolimus in recurrent and refractory pediatric solid tumors, including central nervous system tumors. 2017. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03245151?term=NCT03245151&draw=2&rank=1. Accessed 10 Aug 2017.
Blatt J. Temsirolimus and valproic acid in treating young patients with relapsed neuroblastoma, bone sarcoma, or soft tissue sarcoma. 2010. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01204450?term=NCT01204450&draw=1&rank=1. Accessed 17 Sept 2010.
Ikonomidou C. Chemotherapy and the pediatric brain. Mol Cell Pediatr. 2018;5(1):8.
Watanabe T, et al. Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br J Cancer. 2010;103(10):1580–7.
Milani M, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69(10):4415–23.
Zhu K, et al. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29(3):451–62.
Doherty J, et al. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–7.
Smith M, et al. ER homeostasis and autophagy. Essays Biochem. 2017;61(6):625–35.
Xie C, et al. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy. 2016;12(2):410–23.
Shi W, et al. The role of Atg7-mediated autophagy in ionizing radiation-induced neural stem cell damage. Gene. 2020;738:144485.
Wang Y, et al. Inhibition of autophagy prevents irradiation-induced neural stem and progenitor cell death in the juvenile mouse brain. Cell Death Dis. 2017;8(3):e2694.
Wang Y, et al. Selective neural deletion of the Atg7 gene reduces irradiation-induced cerebellar white matter injury in the juvenile mouse brain by ameliorating oligodendrocyte progenitor cell loss. Front Cell Neurosci. 2019;13:241.
Acknowledgements
Declared none
Funding
Open access funding provided by University of Gothenburg. This work was supported by the National Natural Science Foundation of China (82003396), the Swedish Cancer Foundation (CAN2017/509, 20-1121-PjF), the Swedish Childhood Cancer Foundation (PR2018-0082; PR2021-0020),the Henan Medical Science and Technique Foundation (212102310221, LHCJ20190337), the Henan Provincial Science and Technology Research Project (222102310431), the national Health Commission Key Laboratory of Birth Defects Prevention and Henan Key Laboratory of Population Defects Prevention (2021-03).
Author information
Authors and Affiliations
Contributions
CZ devised the review. YW and YX reviewed the literatures and wrote the manuscript drafts. YW and CZ substantially contributed to the literature review and the writing of this manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuro-oncology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Xu, Y. & Zhu, C. The Role of Autophagy in Childhood Central Nervous System Tumors. Curr. Treat. Options in Oncol. 23, 1535–1547 (2022). https://doi.org/10.1007/s11864-022-01015-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-022-01015-6