
Opinion statement
Advanced colorectal cancer (CRC) is a heterogeneous disease, characterized by several subtypes with distinctive genetic and epigenetic patterns. During the last years, immune checkpoint inhibitors (ICIs) have revamped the standard of care of several tumors such as non-small cell lung cancer and melanoma, highlighting the role of immune cells in tumor microenvironment (TME) and their impact on cancer progression and treatment efficacy. An “immunoscore,” based on the percentage of two lymphocyte populations both at tumor core and invasive margin, has been shown to improve prediction of treatment outcome when added to UICC-TNM classification. To date, pembrolizumab, an anti-programmed death protein 1 (PD1) inhibitor, has gained approval as first-line therapy for mismatch-repair-deficient (dMMR) and microsatellite instability-high (MSI-H) advanced CRC. On the other hand, no reports of efficacy have been presented in mismatch-repair-proficient (pMMR) and microsatellite instability-low (MSI-L) or microsatellite stable (MSS) CRC. This group includes roughly 95% of all advanced CRC, and standard chemotherapy, in addition to anti-EGFR or anti-angiogenesis drugs, still represents first treatment choice. Hopefully, deeper understanding of CRC immune landscape and of the impact of specific genetic and epigenetic alterations on tumor immunogenicity might lead to the development of new drug combination strategies to overcome ICIs resistance in pMMR CRC, thus paving the way for immunotherapy even in this subgroup.
Similar content being viewed by others

Introduction: the role of immune checkpoint inhibitors in dMMR CRC
MLH1, MSH2, MSH6, PMS2, and EPCAM genes encode for proteins involved in DNA mismatch repair (MMR). Alterations of at least one of these genes make identification and repair of spontaneous mutations impossible, leading to rapid accumulation of other variants, high microsatellite instability (MSI-H), and increased tumor mutational load [1,2,3]. Therefore, according to their mutational status, colorectal cancer (CRC) can be divided into two groups: mismatch-repair-deficient (dMMR) and MSI-H tumors (MSI-H-dMMR CRC) and mismatch-repair-proficient (pMMR) and microsatellite instability-low (MSI-L)/microsatellite stable (MSS) tumors (MSI-L/MSS-pMMR CRC). In particular, approximately 15% of all CRC has a MSI-H-dMMR signature [4], and only 3–4% of patients with metastatic CRC (mCRC) detains MSI-H-dMMR status, due to germline, somatic, or epigenetic inactivation MMR genes [1]. In 2015, Le et al. showed that pembrolizumab, a programmed cell death protein 1 (PD1) inhibitor, had a different activity in CRC based on MMR status; the immune-related objective response rate (ORR) was 40%, and progression-free survival (PFS) rate was 78% in MSI-H-dMMR CRC and 0% and 11% respectively for MSI-L/MSS-pMMR CRC [5]. Investigators in the KEYNOTE-177 (a phase 3, open-label trial) compared the efficacy of first-line pembrolizumab monotherapy vs standard chemotherapy in 307 patients affected by MSI-H-dMMR mCRC: Pembrolizumab was superior to chemotherapy in terms of PFS (median 16.5 months vs 8.2 months) (p = 0.0002) and ORR (43.8% vs 33.1%) [6••]. In 2017, the Checkmate-142 trial assessed the activity of first-line monotherapy with nivolumab, another anti-PD1 inhibitor, in 74 patients affected by MSI-H-dMMR mCRC. At a median follow-up of 12 months, the ORR was 31% and 69% of patients detained disease control for 12 weeks or longer [7]. In the same trial, the combination of nivolumab with ipilimumab, a cytotoxic T lymphocyte antigen 4 (CTLA4) inhibitor, achieved 64% ORR, 9% complete response (CR) rate, and 84% disease control rate (DCR) as first-line treatment for MSI-H-dMMR mCRC patients [8, 9]. Therefore, immune checkpoint inhibitors (ICIs) demonstrated particular efficacy in dMMR mCRC but disappointing results in pMMR [5]. The explanation to that could be found in many mechanisms of resistance to immunotherapy such as the presence of immunosuppressive factors in local tumor microenvironment (TME), downregulation of major histocompatibility complex (MHC) protein expression on tumor cell surface, tumor clonal heterogeneity, and tumor dedifferentiation and stemness [10]. Combination strategies aimed to restore tumor immunogenicity are being developed to overcome immunotherapy resistance [11]. To this regard, similarly to other cancer types [12,13,14], prognostic and predictive markers beyond MSI profile are needed, to better identify mCRC patents that could benefit from immunotherapy alone or from drug combination [15,16,17]. The aim of this review is to describe the biological mechanisms of resistance to immune checkpoint inhibitors (ICIs) in MSI-H-pMMR CRC and the therapeutic combination strategies to overcome them.
Biology beyond immune checkpoint inhibitors resistance in pMMR CRC
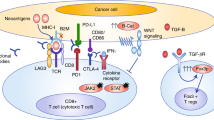
Efficacy of ICIs is related to a wide spectrum of factors, including tumor neoantigens level and presentation, immune cell infiltration and phenotype, and regulatory checkpoint receptors. On the one hand, dMMR CRC harbors a high tumor mutational burden (TMB) that leads to high mutation-associated neoantigen load, higher CD8+ cytotoxic T and Th1 helper cells infiltration [15], and high levels of human leukocyte antigen (HLA) proteins. Conversely, pMMR CRC is considered “immune-excluded” and “cold” tumor [18]. Based on T cell density in each tumor spatial compartment (tumor core, inner invasive margin, and outer invasive margin), tumors are defined “cold” when low T cell density is observed in every compartment, “excluded” when high T cell infiltration is present only in the outer invasive margin, and is low in other compartments and “hot” when high T cell density is present both in the core and inner invasive margin [19••]. ICIs efficacy implies direct contact between tumor-infiltrating lymphocytes (TILs), antigen-presenting cells (APCs), and tumor cells to trigger a strong and specific antitumor immune response. This happens in dMMR CRC, which can be defined as a “hot tumor.” Considering the four consensus molecular subtypes (CMS) [16, 20], each with distinct molecular and immune features, dMMR CRC represents roughly 80% of the CMS1 subgroup; this is characterized by hypermutated phenotype and strong immune activation. Conversely, the immunogenic scenario changes radically in the other subgroups, shifting toward a less immunogenic TME (Fig. 1). CMS2, CMS3, and CMS4 subtypes include mainly pMMR CRC. CMS2 subtype includes 35% of CRC and is characterized by chromosomal instability (CIN) and upregulation of WNT and MYC pathways: This subtype might be defined as an “immune-excluded tumor.” The same might be said for CMS3 subtype, characterized by a high rate of K-RAS mutations and usually defined as the metabolic subgroup [21]. Recent studies have shown that CMS2 and CMS3 detain a poor immunogenic TME, with low levels of TILs and APCs inside tumor core. Therefore, in these CRC subtypes, ICIs monotherapy seems to be ineffective. A work of Jason JL et al., using The Cancer Genome Atlas (TCGA) tumor samples to evaluate a T cell-inflamed gene expression signature within different tumor types, demonstrated that tumors with activation of WNT/b-catenin signaling were characterized by a non-T cell-inflamed TME [22]. In preclinical murine models, a suppressive activity on CCL4 gene transcription by WNT/β-catenin pathway, commonly upregulated in CMS2/pMMR CRC tumors, was observed, leading to low levels of CCL4 chemokine and impaired CD103+ dendritic cells (DCs) and CD8+ T cell infiltration and subsequent activation in TME [23, 24]. Somatic mutations of K-RAS or N-RAS involve about 60% of CRC, mostly pMMR tumors, leading to a constitutive activation of the mitogen-activated protein kinase (MAPK) pathway [25]. Researchers demonstrated that KRAS mutations, common in CMS3 CRC subtype, promote an immunosuppressive TME, inducing the conversion of CD4+ cells to Tregs [26, 27•] and, by upregulation of CXCL3 expression, the main ligand of CXCR2 on MDSCs surface, support their migration inside tumor core [28]. Moreover, KRAS activation reduces tumor immunogenicity, downregulating MHC-I molecules and leading to inability of CD8+ T cells to recognize tumor cells [27•]. CMS4 subgroup, also known as “mesenchymal subgroup,” includes only 6% dMMR samples and is therefore largely represented by pMMR CRC and is characterized by strong stromal activity, angiogenesis, and TGF-β pathway activation. These tumors detain an inflamed TME, enriched of immune cells with an immunosuppressive activity such as M2 macrophages, regulatory T cells (Treg), and myeloid-derived suppressor cells (MDSCs), leading to ICIs resistance. TGF-β is involved in the control of adaptive immunity leading to the expansion of Treg and inhibition of effector T cells [29, 30]. The TGF-β pathway suppresses antigen presentation of DCs by inhibiting IFN-γ-mediated induction of the class II transactivator (CIITA) promoter, essential for MHC-II gene expression [31]. TGF-β is mostly produced by cancer-associated fibroblasts (CAFs), a group of stromal cells involved in the production of extracellular matrix components, such as collagens and fibronectin, as well as several cytokines that regulate cancer progression. In this subgroup of pMMR CRC, the higher TGF-β levels produced by CAFs lead to the exclusion of CD4+ and CD8+ T cells from the tumor center and, therefore, to ICIs inefficacy [32]. Mariathasan et al. demonstrated that the immune-excluded phenotype might be overcome using anti-Pan-TGF-b antibody, promoting T cell priming and concentration in tumor core and restoring anti PD-L1 treatment efficacy in mouse tumor models [33•]. Response to ICIs implies not only the infiltration of immune cells with a certain phenotype inside TME but also the presentation of neoantigens on tumor cells to be recognized by T cells. TMB represents the number of somatic mutations per coding area of a tumor genome and has been proposed as a predictor of response to ICIs monotherapy [34]. In this context, pMMR CRC, with a median TMB of 4 mutations/MB, is defined as a low TMB tumor [35•], with a poor load of cancer specific antigens to be detected by cytotoxic CD8+ T cells, further explaining ICIs inefficacy in this CRC subgroup [18, 36].

pMMR CRC are characterized by an immune-excluded and immune-suppressive tumor microenvironment (TME), leading to resistance to immune checkpoint inhibitors (ICIs). In fact, tumor-infiltrating lymphocytes (TILs) and antigen-presenting cells (APCs) are located outside of tumor core and inner invasive margin, reducing their direct contact with tumor cells. Inside tumor core, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Treg) lead to suppression of immune response against cancer cells. Moreover, pMMR CRC detains low levels of neoantigens, impairing their presentation to CD8+ T cells. In this immune-excluded and immune-suppressive TME, chemotherapy, inducing immunogenic cell death (ICD), promotes exposure on tumor cell surface of calreticulin, able to inhibit tumor angiogenesis and increased TILs inside TME, and of heat shock protein (HSP) 70 and 90, able to bind CD40 on dendritic cells (DCs) and activating them. Moreover, ICD promotes the release from tumor cells of HMGB1 that could bind TLR4 on DCs surface and promote their activation. Radiotherapy leads to increased neoantigen presentation and release from tumor cells and increased expression of PD-L1. Anti-angiogenesis drugs, such as bevacizumab, and other multikinase inhibitors (MKI), such as regorafenib, are able to inhibit the immuno-suppressive effect of VEGF/VEGFR on TME, blocking the infiltration of MDSCs, TAMs, and Treg inside TME and the expression of T cell exhaustion markers (PD-1, LAG3, TIM3). Anti-EGFR drugs, inducing antibody-dependent cell-mediated cytotoxicity (ADCC), lead to cancer cells lysis by natural killer (NK) cells. Finally, alterations of MAPK pathway on cancer cells promote migration of suppressive immune cells inside TME and reduce TILs infiltration; therefore, drugs that target this axis might restore antitumor immune cell activity.
Chemotherapy and immune checkpoint inhibitors combination
Standard chemotherapy represents the mainstay of treatment for the majority of CRC patients, either in resected stage II or III [37], and also for stage IV where polychemotherapy with 5-fluorouracil (5FU) and irinotecan, oxaliplatin, or both [38] is widely used in combination with either anti-VEGF or anti-EGFR drugs. ICIs in unselected mCRC patients usually yield poor results [39]: Since MSI-L/MSS CRC is inherently resistant to ICIs, most MSI-H patients who exhibited primary resistance to ICIs were mainly due to MSI/MMR misdiagnoses more than any other factor [40]. In lung cancer patients, a combination of ICIs and chemotherapy has been developed, improving prognosis in patients who seemed to respond poorly to ICIs monotherapy [41, 42]. Indeed, chemotherapeutic agents seem to detain immunostimulatory properties [43], and this combination strategy is currently under investigation in other tumor types [44], such as CRC. Roselli et al. [45] reported changes of peripheral blood mononuclear cell count in patients treated with FOLFIRI + bevacizumab after treatment; CD4+ T cells were increased, and Treg ratios were decreased as well. In patients where Treg decrease was observed, statistically significant better OS (p = 0.036), PFS (p = 0.037), and ORR (p = 0.0064) were seen. Van Der Kraak [46] reported that human CRC HT-116 cell lines, treated with 5FU, had a statistically significant increase in PD-L1 levels after treatment. Oxaliplatin can induce tumor cell death through a mechanism called immunogenic cell death (ICD) [47]. Therefore, its potential role as immunomodulatory agent has been widely investigated in CRC. Tesniere et al. [48] reported that oxaliplatin in CRC murine models lead to the exposure of calreticulin on membrane surface and release of high-mobility group box 1 protein (HMGB1), able to bind TLR4 of DCs and activate them [49]. Moreover, calreticulin, in addition to inhibiting angiogenesis, seems to increases TILs in TME, enhancing ICIs activity [50]. Song et al. [51] investigated the efficacy of oxaliplatin and anti-PD-L1 agent in a murine CRC model, and they showed that when oxaliplatin was added to anti-PD-L1 drug, a statistically significant reduction in tumor growth was observed compared to oxaliplatin alone. Both studies used CT26 cell line, a renowned MSS, K-RAS G12D mutated cell line [52].
Wang et al. [53] compared different sensitivity to ICIs-oxaliplatin combination in different cell lines: CT26, sensitive to ICIs, vs MC38, resistant to ICIs. This study demonstrated that MC38 cells also “regained” their sensitivity to ICIs treatment after oxaliplatin exposure. Golchin et al. [54••] also reported that oxaliplatin in combination with anti-PD-L1 was associated with improved survival and tumor control compared with oxaliplatin or anti-PD-L1 alone in CT26 tumor-bearing mice. There are several ongoing/completed trials that aim to assess the impact of chemotherapy as an immune-sensitizing agent in CRC (Table 1). The phase II POCHI trial (NCT04262687) will assess the impact of chemotherapy and ICIs in MSS CRC patients, stratified by immune cells infiltrate. Patients will receive XELOX + bevacizumab + pembrolizumab combination. Immunoscore and TuLIS score will also be performed at baseline as to evaluate their role as predictors of response to ICIs in MSS CRC patients. Finally, another phase I study (NCT03626922) will assess the impact of combination of pembrolizumab + oxaliplatin + pemetrexed in patients with MSS CRC. The study is currently ongoing.
Radiotherapy and immune checkpoint inhibitors combination
Radiotherapy is rarely used in mCRC, mainly as palliative treatment of bone metastases. On the other hand, chemoradiation (CRT) is standard of treatment of locally advanced rectal cancer, either in neoadjuvant or adjuvant setting. A few papers have suggested that rectal cancer CRT might determine an increase in PD-L1 expression, thus potentially improving ICIs efficacy in this disease. Hecht et al. [55] compared PD-L1 expression between 103 pre-CRT biopsies and 159 post-CRT surgical specimens. Authors confirmed increased PD-L1 expression after CRT, particularly in the cancer invasive front (from 2.1 to 9.3%, p < 0.001). Low PD-L1 expression and low immune cells expression were associated with worse OS. Chen et al. [56] came to similar results: In 112 matched pre- and post-CRT locally advanced rectal cancer (LARC) biopsies, PD-L1 expression levels and tumor-infiltrating CD8+ T cells were increased after CRT. Pre- and post-CRT levels of both PD-L1 and CD8+ T-cells were associated with improved survival and reduced relapse risk. On the other hand, Shao et al. [57] reported that in patients treated with CRT, high PD-L1 expression was associated with statistically significant worse OS and higher local relapse rate, suggesting that these patients would require additional anti-PD-L1 therapy. Saigusa et al. [58] also reported that in 90 LARC patients treated with CRT, higher levels of PD-L1 levels were associated with higher risk of locoregional relapse (p = 0.0051). Higher levels of PD-L1 expression were associated with lower CD8+ lymphocytes. All these papers seem to suggest that CRT in LARC might cause an increase in expression of PD-L1 and also TILs in TME. On the other hand, the prognostic role of higher PD-L1 expression, particularly after CRT, seems to be less defined. As for clinical trials that will assess the real impact of radiotherapy and ICIs, a series of ongoing trials are actually been conducted (Table 1).
Anti-VEGF and immune checkpoint inhibitors combination
There are a few published papers showing close relationship between VEGF (vascular endothelial growth factor)-driven angiogenesis and immune TME, suggesting the use of anti-VEGF treatment in combination with ICIs to overcome resistance in pMMR CRCs. Preclinical studies demonstrated that VEGF-driven angiogenesis leads to the expansion of suppressive immune cells including Tregs and MDSCs [59, 60] and increases tumor-associated macrophages’ (TAMs) infiltrates in tumor sites [61]. On the other hand, VEGF exerts its immunosuppressive effect also by the inhibition of the progenitor cells differentiation to CD4+ and CD8+ lymphocytes [62] with T cells decreased proliferation and reduced cytotoxic effects. In addition to that, VEGF has been shown to increase T cell exhaustion by increasing PD-1, CTLA-4, TIM3, and LAG3 expression on T cells. Interestingly, VEGF immunosuppressive effects have been shown to be reversible with the use of anti-VEGF drugs, providing a strong rationale for the combination of angiogenesis inhibitors and ICIs. Bevacizumab, an anti-VEGF-A antibody commonly used for mCRC treatment in combination with chemotherapy, has been evaluated in combination with ICIs in CRC in different clinical trials with conflicting results. In the phase II BACCI trial, the addition of atezolizumab to a combination of capecitabine and bevacizumab was assessed in a cohort of refractory metastatic CRC patients, providing a modest improvement in PFS (4.4 vs 3.3 months, HR = 0.72; p = 0.051) [63]. Another study, the MODUL trial, evaluated the addition of atezolizumab to maintenance therapy with 5FU and bevacizumab after and induction with FOLFOX and bevacizumab in patients with pMMR CRC [64] with no evidence of benefit in terms of PFS or OS. The combination of ICIs with bevacizumab and chemotherapy in first-line setting was assessed in a phase Ib trial where atezolizumab was associated with FOLFOX and bevacizumab regimen [65]. The study showed no unexpected safety signals and encouraging results in term of activity with a median PFS of 14.1 months (95% CI 8.7–17.1) and a median duration of response of 11.4 months (95% CI 7.6–15.9). The authors also demonstrated that CD8+ T-cells and PD-L1 expression were increased in tumors following administration of FOLFOX, atezolizumab, and bevacizumab. Moreover, patients with elevations in tumor-infiltrating CD8+ T cells consistent with increased expression of cytotoxic T cell signatures and PD-L1 showed sustained responses or prolonged disease control, confirming that the hypothesis that this combination could promote immune-related antitumoral activity. The use of an intensified chemotherapy regimen, such as FOLFOXIRI, in combination with bevacizumab and ICIs, is currently under evaluation in two clinical trials (ATEZOTRIBE and NIVACOR). The rationale behind this strategy relies in the observation that intensification of the chemotherapy plus bevacizumab can boost the release of novel neoantigens and infiltration of CD8+ T cells, increasing the likelihood of response to immunotherapy. Preliminary results of phase II NIVACOR trial have been recently presented [66]. The study includes patients with RAS or BRAF mutated, regardless of microsatellite status, mCRC patients treated with FOLFOXIRI and bevacizumab in association with nivolumab for eight cycles followed by maintenance treatment with bevacizumab and nivolumab. The combination was generally well tolerated with an acceptable toxicity profile and no unexpected findings. The ATEZOTRIBE is a randomized phase II trial comparing FOLFOXIRI/bevacizumab with FOLFOXIRI/bevacizumab plus atezolizumab in first-line treatment of mCRC (NCT03721653). As in the NIVACOR trial, after the induction phase, patients are treated with maintenance treatment with 5-FU and bevacizumab or the same combination in association with atezolizumab. Primary endpoint of the study is PFS (Table 1).
Anti-EGFR drugs and immune checkpoint inhibitors combination
Cetuximab and panitumumab, monoclonal antibodies targeting epidermal growth factor receptor (EGFR), are currently used for the systemic treatment of mCRC in combination with standard chemotherapy in RAS and BRAF wild-type (wt) mCRC [67, 68]. During the last years, preclinical works have shown that immunoglobulin (Ig) G1 monoclonal antibodies (mAbs) detain a high capability for stimulating antibody-dependent cell-mediated cytotoxicity (ADCC) [69, 70•]. ADCC is an immune mechanism involving the killing of antibody-coated target cells expressing tumor-derived antigens on their surface by effector cells, usually natural killer (NK) cells [71]. Cetuximab, binding to EGFR on cancer cells and to the CD16 receptor on NK and DCs, has been shown to induce ADCC and, therefore, the secretion of pro-inflammatory cytokines (i.e., IFN-γ, TNFα) and the priming of cytotoxic T cells in the TME, stimulating immunity against tumor [72,73,74]. However, this initial immune stimulation is followed by the induction of immuno-suppressive mechanisms such as recruitment of Treg and MDSCs on TME and increased expression of PD-1, PD-L1, and CTLA-4 on tumor and immune cells. The synergism between anti-EGFR and ICIs might be expected on the basis of a two-step process where anti-EGFR drugs contribute to immune cell recruitment in TME and ICIs allow reactivation of immune cells that are already present; this might lead to forefront combinations in order to overcome ICIs resistance in pMMR CRC—RAS wt tumors [75]. An ongoing phase I/II trial (NCT02713373) is evaluating cetuximab and pembrolizumab combination in RAS wt mCRC, after at least one prior treatment line. Remarkably, 6 out of 9 patients achieved stable disease (SD) lasting ≥ 16 weeks, with no dose limiting toxicities (DLTs) observed [76]. The AVATUX study (NCT03174405), a phase II trial, investigated the combination of avelumab, chemotherapy (mFOLFOX6 regimen) and cetuximab in first-line RAS and BRAF wt mCRC. Preliminary results on 43 patients showed encouraging data [77]. Specifically, ORR was 79.5%, including 6 CR and 25 partial responses (PR). Moreover, 5 SD were noted; thus, DCR was 92.3%.
Phase II trial (NCT03442569) combining nivolumab and ipilimumab with panitumumab in RAS and BRAF wt pretreated MSS mCRC met its primary endpoint with a 12-week response rate of 35% [78]. In addition to these data, two clinical trials are ongoing: the AVETUXIRI trial (NCT03608046), which is investigating the efficacy of avelumab combined with cetuximab and irinotecan in patients with refractory mCRC, and the CAVE colon (2017-004392-32), a phase II study designed to evaluate the efficacy of avelumab and cetuximab in pretreated RAS WT mCRC patients (Table 1).
MAPK pathway inhibitors and immune checkpoint inhibitors combination
MAP kinase pathway is crucial in tumor initiation and progression, and K-RAS and BRAF mutations are detected in around 30–50% and 5–10 % of CRC, respectively [79, 80]. Recent studies demonstrated in mCRC patients that molecular alterations of MAPK pathway have also immunosuppressive properties. KRAS mutations in cancer cells detain autocrine actions and impact on TME components, promoting an immunosuppressive stroma through the induction of cytokines and growth factors’ secretion [27•, 81•]. Moreover, KRAS mutations lead to upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) in TME of CRC, enhancing the infiltration of MDSCs [82] and causing an evasion of antitumor immunity. MAPK pathway is involved in the regulation of HLA I expression, and its inhibition leads to the upregulation of HLA I molecules on cancer cell surface, facilizing recognition by CD8+ T cell [83,84,85]. Moreover, its activation on cancer cells reduces TILs, leading to tumor cell immune evasion [85, 86]. Before the discovery of K-RAS G12C isoform inhibitors AMG510 (sotorasib) and MRTX849 (adagrasib), K-RAS mutations had been considered undruggable [79]. A recent preclinical study showed the immunomodulatory properties of KRAS G12C inhibitors: After treatment with AMG510, an increased infiltration of T cells, primarily CD8+ T cells, into KRAS G12C mutated tumors in rats was observed [87••].
Phase 1/1b AMG510 CodeBreak 100 [88••] and MRTX849 Kristal-1 [89] ongoing trials have shown promising results, respectively, providing over 50 % and 90% DCR in heavily pretreated patients with KRAS G12C mutated solid tumors. Therefore, sotorasib is being studied in combination with MEK inhibitors or anti-PD-1 agents in NSCLC and CRC in CodeBreak101 [90], while Kristal-7 will provide data about adagrasib and pembrolizumab doublet in NSCLC. Even though K-RAS G12C variant is present in only 10% of all K-RAS mutations in CRC patients [79], the results of these trials might be crucial in a clinical setting where other treatment options are rather limited. Focusing on RAF/MEK axis, the combination of encorafenib + binimetinib + nivolumab and dabrafenib + trametinib + spartalizumab, an anti-PD1 drug, in patients affected by BRAF-V600E mutated pMMR CRC is being evaluated in phase I/II NCT04044430 and phase II NCT03668431 trials respectively. Preliminary data have reported 33% ORR of both combinations. Moreover, phase I/II NCT03711058 is assessing PIK3CA inhibition with copanlisib combined with nivolumab in relapsed/refractory solid tumors, including MSS CRC. Preliminary results of IMblaze370 [91], a phase III randomized trial comparing atezolizumab plus cobimetinib, a MEK1/2 inhibitor vs atezolizumab monotherapy vs regorafenib in third-line setting in pMMR CRC, are less promising. Atezolizumab + cobimetinib regimen had 8.87 months mPFS vs 7.10 months for atezolizumab alone vs 8.51 months for regorafenib. Moreover, the combined treatment arm with atezolizumab + cobimetinib was associated with a significantly higher (64%) grade 3–4 adverse events’ rate compared with the other study arms (Table 1).
Multi-target kinase inhibitors and immune checkpoint inhibitors combination
TAMs are involved in cancer progression inducing angiogenesis through VEGF secretion, immune evasion through immunosuppressive cytokines release (IL-10, TGFβ), and upregulating immune checkpoint expression such as PD-L1 on tumor cells [92,93,94,95]. Regorafenib is a multi-target kinase inhibitor (MKI) that not only inhibits VEGF and its receptor VEGFR but also other pro-angiogenetic molecules, highly expressed by TAMs, such as EGF homology domain 2 (TIE2), blocking their recruitment into the TME [96]. Therefore, combinations of ICIs and regorafenib or lenvatinib, another MKI, are currently under investigation. Regorafenib plus nivolumab combination [97] showed 40% ORR in the phase Ib REGONIVO study in both gastric and colorectal cancer cohorts. Thirty-six percent of ORR was observed in the CRC cohort alone with 7.9 mPFS in pMMR tumors. Phase II REGOMUNE trial [98] with a regorafenib and avelumab combination reported 53.3% SD rate; however, no objective responses were seen. Although median PFS and median OS were quite modest (3.6 and 10.8 months respectively), pathological review of tumor samples showed that a significant increase in CD8+ T cells infiltrates in 60% of cases and this change was associated with improved outcome, suggesting a synergism in shaping the TME of this drug combination. Conversely, higher baseline TAMs infiltration was significantly associated with shorter PFS. Combination of ICIs and lenvatinib (NCT03797326) is under investigation in a phase II study in patients with previously treated advanced solid tumors, including pMMR CRC. This study was based also on the results of the LEMON trial [99], a phase II study that showed 70% DCR rate of lenvatinib monotherapy in patients with mCRC refractory to standard chemotherapy. Another phase Ib/II trial (NCT02713529) will assess the combination of pembrolizumab with AMG 820, an antibody targeting colony-stimulating factor 1 receptor (CSF-1R) that regulates TAMs recruitment and survival; preliminary data of this study have reported only 4.9% ORR in pMMR CRC patients’ cohort (Table 1).
Conclusions
After the discovery of the effectiveness of ICIs in several tumor types such as melanoma, lung cancer, and kidney cancer, researchers have investigated the activity of these drugs also other solid tumors. In gastrointestinal malignancies, most studies have been met with disappointment, with the notable exception of patients with MSI-H-dMMR subset. As it stands, approximately 95% of mCRC patients, represented by those with pMMR mCRC tumors, remain outside from this promising therapeutic opportunity. On the second thought, lack of activity of ICIs monotherapy in majority of mCRC should not be surprising, based on the wide genetic and epigenetic heterogeneity associated with different types of CRC. Starting from CMS classification, as previously discussed, each CMS detains its own molecular identity from which derives the immunogenicity of the tumor itself. As it can be seen, most promising ways to introduce ICIs in clinical setting for majority of patients with mCRC are those where these drugs will be combined with other treatment options. Most trials have focused on biological rationale of new drug combinations that might turn what is usually considered from an immunogenic point of view as “cold” tumor into a “hot” tumor.
What seems to be lacking in all these trials is a prospectively defined translational research assessment (with a few notable exceptions): It is eagerly awaited that based on the innovative mechanism of action of ICIs compared with standard chemotherapy, previous “errors” that were committed during the development of anti-VEGF and anti-EGFR-based therapy might be overcome by taking into account pre-specified molecular stratification of these patients.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol Mech Dis. 2011;6:479–507. https://doi.org/10.1146/annurev-pathol-011110-130235.
Amin M, Lockhart AC. The potential role of immunotherapy to treat colorectal cancer. Expert Opin Investig Drugs. 2015;24:329–44. https://doi.org/10.1517/13543784.2015.985376.
Ghidini M, Fusco N, Salati M, Khakoo S, Tomasello G, Petrelli F, et al. The emergence of immune-checkpoint inhibitors in colorectal cancer therapy. Curr Drug Targets. 2021;22. https://doi.org/10.2174/1389450122666210204204415.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16:361–75. https://doi.org/10.1038/s41575-019-0126-x.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. https://doi.org/10.1056/nejmoa1500596.
•• André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. 2020. https://doi.org/10.1056/nejmoa2017699 This reference is of outstanding importance because is the phase III trial that demonstrated the superiority of first line pembrolizumab in metastatic MSI-H-dMMR colorectal cancer compared to standard chemotherapy, leading to significantly longer progression-free survival.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. https://doi.org/10.1016/S1470-2045(17)30422-9.
Lenz H-J, Lonardi S, Zagonel V, Van Cutsem E, Limon ML, Wong KYM, et al. Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite instability-high/DNA mismatch repair deficient metastatic colorectal cancer: clinical update. J Clin Oncol. 2020;38:11. https://doi.org/10.1200/jco.2020.38.4_suppl.11.
Pernot S, Terme M, Voron T, Colussi O, Marcheteau E, Tartour E, et al. Colorectal cancer and immunity: what we know and perspectives. World J Gastroenterol. 2014;20:3738–50. https://doi.org/10.3748/wjg.v20.i14.3738.
Almquist DR, Ahn DH, Bekaii-Saab TS. The role of immune checkpoint inhibitors in colorectal adenocarcinoma. BioDrugs. 2020;34:349–62. https://doi.org/10.1007/s40259-020-00420-3.
Kreidieh M, Mukherji D, Temraz S, Shamseddine A. Expanding the scope of immunotherapy in colorectal cancer: current clinical approaches and future directions. Biomed Res Int. 2020;2020:1–24. https://doi.org/10.1155/2020/9037217.
Cortellini A, Tiseo M, Banna GL, Cappuzzo F, Aerts JGJV, Barbieri F, et al. Clinicopathologic correlates of first-line pembrolizumab effectiveness in patients with advanced NSCLC and a PD-L1 expression of ≥50%. Cancer Immunol Immunother. 2020;69:2209–21. https://doi.org/10.1007/s00262-020-02613-9.
Cantini L, Pecci F, Hurkmans DP, Belderbos RA, Lanese A, Copparoni C, et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur J Cancer. 2021;144:41–8. https://doi.org/10.1016/j.ejca.2020.10.031.
Cortellini A, Ricciuti B, Tiseo M, Bria E, Banna GL, Aerts JGJV, et al. Baseline BMI and BMI variation during first line pembrolizumab in NSCLC patients with a PD-L1 expression ≥50%: A multicenter study with external validation. J Immunother Cancer. 2020;8:e001403. https://doi.org/10.1136/jitc-2020-001403.
Huyghe N, Baldin P, Van Den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep. 2020;8:11–24. https://doi.org/10.1093/gastro/goz061.
Guinney J, Dienstmann R, Wang X, De Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. https://doi.org/10.1038/nm.3967.
Giampieri R, Cantini L, Giglio E, Bittoni A, Lanese A, Crocetti S, et al. Impact of polypharmacy for chronic ailments in colon cancer patients: a review focused on drug repurposing. Cancers (Basel). 2020;12. https://doi.org/10.3390/cancers12102724.
Ghiringhelli F, Fumet JD. Is there a place for immunotherapy for metastatic microsatellite stable colorectal cancer? Front Immunol. 2019;10. https://doi.org/10.3389/fimmu.2019.01816.
•• Pai SI, Cesano A, Marincola FM. The paradox of cancer immune exclusion: immune oncology next frontier. Cancer Treat Res. 2020. https://doi.org/10.1007/978-3-030-38862-1_6 This reference is of outstanding importance because clearly explains the various possible scenarios of TME and how immune cells might be placed inside TME, leading to different responses to ICIs.
Bittoni A, Sotte V, Meletani T, Cantini L, Giampieri R, Berardi R. Immunotherapy in colorectal cancer treatment: actual landscape and future perspectives. J Cancer Metastasis Treat. 2018:10.20517/2394–4722 2018.37.
Becht E, De Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22:4057–66. https://doi.org/10.1158/1078-0432.CCR-15-2879.
Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/b-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25:3074–83. https://doi.org/10.1158/1078-0432.CCR-18-1942.
Williford JM, Ishihara J, Ishihara A, Mansurov A, Hosseinchi P, Marchell TM, et al. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci Adv. 2019;5:eaay1357. https://doi.org/10.1126/sciadv.aay1357.
Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–723.e4. https://doi.org/10.1016/j.ccell.2017.04.003.
Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–27. https://doi.org/10.1038/nrclinonc.2009.111.
Zdanov S, Mandapathil M, Eid RA, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res. 2016;4:354–65. https://doi.org/10.1158/2326-6066.CIR-15-0241.
• Hamarsheh S, Groß O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun. 2020. https://doi.org/10.1038/s41467-020-19288-6 These reference are of importance because clearly takes stock of the impact of Kras mutations in cancer cells on tumor immunogenicity and immune cells phenotype and activity inside TME.
Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35:559–572.e7. https://doi.org/10.1016/j.ccell.2019.02.008.
Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50:924–40. https://doi.org/10.1016/j.immuni.2019.03.024.
Sanjabi S, Oh SA, Li MO. Regulation of the immune response by TGF-β: from conception to autoimmunity and infection. Cold Spring Harb Perspect Biol. 2017;9. https://doi.org/10.1101/cshperspect.a022236.
Piskurich JF, Wang Y, Linhoff MW, White LC, Ting JP. Identification of distinct regions of 5’ flanking DNA that mediate constitutive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. J Immunol. 1998.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–43. https://doi.org/10.1038/nature25492.
• Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018. https://doi.org/10.1038/nature25501 This reference is of importance beacuse clearly explains the immunosuppressive effect of TGFβ signalling in stromal cells on TME, leading to an "immune-excluded" phenotype and resistance to immunotherapy.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598–608. https://doi.org/10.1158/1535-7163.MCT-17-0386.
• Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017. https://doi.org/10.1186/s13073-017-0424-2 This references is of importance because this work is an overview of the TMB among many solid cancer types and highlights the relationship between high TMB and microsatellite instability status.
Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25:89–94. https://doi.org/10.1038/s41591-018-0266-5.
André T, Meyerhardt J, Iveson T, Sobrero A, Yoshino T, Souglakos I, et al. Effect of duration of adjuvant chemotherapy for patients with stage III colon cancer (IDEA collaboration): final results from a prospective, pooled analysis of six randomised, phase 3 trials. Lancet Oncol. 2020;21:1620–9. https://doi.org/10.1016/S1470-2045(20)30527-1.
Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrine G, Barbara C, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: The gruppo oncologico nor. J Clin Oncol. 2007;25:1670–6. https://doi.org/10.1200/JCO.2006.09.0928.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;80. https://doi.org/10.1126/science.aan6733.
Cohen R, Hain E, Buhard O, Guilloux A, Bardier A, Kaci R, et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol. 2019;5:551–5. https://doi.org/10.1001/jamaoncol.2018.4942.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N Engl J Med. 2018;379:2040–51. https://doi.org/10.1056/nejmoa1810865.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. 2018;378:2078–92. https://doi.org/10.1056/nejmoa1801005.
Opzoomer JW, Sosnowska D, Anstee JE, Spicer JF, Arnold JN. Cytotoxic chemotherapy as an immune stimulus: a molecular perspective on turning up the immunological heat on cancer. Front Immunol. 2019;10. https://doi.org/10.3389/fimmu.2019.01654.
Weiss GJ, Waypa J, Blaydorn L, Coats J, Mcgahey K, Sangal A, et al. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br J Cancer. 2017;117:33–40. https://doi.org/10.1038/bjc.2017.145.
Roselli M, Formica V, Cereda V, Jochems C, Richards J, Grenga I, et al. The association of clinical outcome and peripheral T cell subsets in metastatic colorectal cancer patients receiving first-line FOLFIRI plus bevacizumab therapy. Oncoimmunology. 2016;5:e1188243. https://doi.org/10.1080/2162402X.2016.1188243.
Van Der Kraak L, Goel G, Ramanan K, Kaltenmeier C, Zhang L, Normolle DP, et al. 5-Fluorouracil upregulates cell surface B7-H1 (PD-L1) expression in gastrointestinal cancers. J Immunother Cancer. 2016;4:65. https://doi.org/10.1186/s40425-016-0163-8.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. https://doi.org/10.1146/annurev-immunol-032712-100008.
Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91. https://doi.org/10.1038/onc.2009.356.
Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211. https://doi.org/10.1186/1479-5876-12-211.
Wang HT, Lee HI, Guo JH, Chen SH, Liao ZK, Huang KW, et al. Calreticulin promotes tumor lymphocyte infiltration and enhances the antitumor effects of immunotherapy by up-regulating the endothelial expression of adhesion molecules. Int J Cancer. 2012;130:2892–902. https://doi.org/10.1002/ijc.26339.
Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237. https://doi.org/10.1038/s41467-018-04605-x.
Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. https://doi.org/10.1186/1471-2164-15-190.
Wang W, Wu L, Zhang J, Wu H, Han E, Guo Q. Chemoimmunotherapy by combining oxaliplatin with immune checkpoint blockades reduced tumor burden in colorectal cancer animal model. Biochem Biophys Res Commun. 2017;487:1–7. https://doi.org/10.1016/j.bbrc.2016.12.180.
•• Golchin S, Alimohammadi R, Rostami Nejad M, Jalali SA. Synergistic antitumor effect of anti-PD-L1 combined with oxaliplatin on a mouse tumor model. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.28585 This reference is of outstanding importance because shows the synergism between ICIs and chemotherapy. In fact, combination of Oxaliplatin and anti-PD-L1 significantly increased survival in vivo and inhibited tumor growth of tumor-bearing mice and showed how Oxaliplatin can change tumor microenvironment from immune-suppressive toward the immune-favorable condition.
Hecht M, Büttner-Herold M, Erlenbach-Wünsch K, Haderlein M, Croner R, Grützmann R, et al. PD-L1 is upregulated by radiochemotherapy in rectal adenocarcinoma patients and associated with a favourable prognosis. Eur J Cancer. 2016;65:52–60. https://doi.org/10.1016/j.ejca.2016.06.015.
Chen TW, Huang KCY, Chiang SF, Chen WTL, Ke TW, Chao KSC. Prognostic relevance of programmed cell death-ligand 1 expression and CD8+ TILs in rectal cancer patients before and after neoadjuvant chemoradiotherapy. J Cancer Res Clin Oncol. 2019;145:1043–53. https://doi.org/10.1007/s00432-019-02874-7.
Shao L, Peng Q, Du K, He J, Dong Y, Lin X, et al. Tumor cell PD-L1 predicts poor local control for rectal cancer patients following neoadjuvant radiotherapy. Cancer Manag Res. 2017;Volume 9:249–58. https://doi.org/10.2147/CMAR.S139889.
Saigusa S, Toiyama Y, Tanaka K, Inoue Y, Mori K, Ide S, et al. Implication of programmed cell death ligand 1 expression in tumor recurrence and prognosis in rectal cancer with neoadjuvant chemoradiotherapy. Int J Clin Oncol. 2016;21:946–52. https://doi.org/10.1007/s10147-016-0962-4.
Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T- cells in malignant effusions. Anticancer Res. 2009.
Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. 2007;110:624–31. https://doi.org/10.1182/blood-2007-01-065714.
Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005;15:417–25. https://doi.org/10.1097/00008390-200510000-00010.
Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–86. https://doi.org/10.1182/blood-2002-07-1956.
Mettu NB, Twohy E, Ou F-S, Halfdanarson TR, Lenz HJ, Breakstone R, et al. BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann Oncol. 2019;30:v203. https://doi.org/10.1093/annonc/mdz246.011.
Grothey A, Tabernero J, Arnold D, De Gramont A, Ducreux MP, O’Dwyer PJ, et al. Fluoropyrimidine (FP) + bevacizumab (BEV) + atezolizumab vs FP/BEV in BRAFwt metastatic colorectal cancer (mCRC): findings from Cohort 2 of MODUL – a multicentre, randomized trial of biomarker-driven maintenance treatment following first-line induction th. Ann Oncol. 2018;29:viii714–5. https://doi.org/10.1093/annonc/mdy424.020.
Wallin J, Pishvaian MJ, Hernandez G, Yadav M, Jhunjhunwala S, Delamarre L, et al. Abstract 2651: Clinical activity and immune correlates from a phase Ib study evaluating atezolizumab (anti-PDL1) in combination with FOLFOX and bevacizumab (anti-VEGF) in metastatic colorectal carcinoma; 2016. https://doi.org/10.1158/1538-7445.am2016-2651.
Damato A, Berselli A, Iachetta F, Romagnani A, Larocca M, Garcia Arias A, et al. Preliminary safety analysis of phase II open-label NIVACOR trial (GOIRC-03-2018) in patients with advanced colorectal cancer RAS or BRAF mutated. J Clin Oncol. 2021;39:37. https://doi.org/10.1200/jco.2021.39.3_suppl.37.
Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. https://doi.org/10.1056/nejmoa1305275.
Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezínek I, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33:692–700. https://doi.org/10.1200/JCO.2014.59.4812.
Lo NC. Evaluation of antibody-dependent cell-mediated cytotoxicity activity and cetuximab response in KRAS wild-type metastatic colorectal cancer patients. World J Gastrointest Oncol. 2016;8:222–30. https://doi.org/10.4251/wjgo.v8.i2.222.
• Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med. 2019. https://doi.org/10.21037/atm.2019.01.42 This reference is of importance because takes stock of the role of antibody-dependent cell-mediated cytotoxicity (ADCC), mediated by natural killer cells, in anti tumor activity, leading to the stimulation of adaptive immune response, thus becoming an ally of immune check-point inhibitors.
Zahavi D, AlDeghaither D, O’Connell A, Weiner LM. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antib Ther. 2018;1:7–12. https://doi.org/10.1093/abt/tby002.
Trotta AM, Ottaiano A, Romano C, Nasti G, Nappi A, De Divitiis C, et al. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol Res. 2016;4:366–74. https://doi.org/10.1158/2326-6066.CIR-15-0184.
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. https://doi.org/10.1016/j.ccr.2005.03.003.
Trivedi S, Srivastava RM, Concha-Benavente F, Ferrone S, Garcia-Bates TM, Li J, et al. Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients. Clin Cancer Res. 2016;22:5229–37. https://doi.org/10.1158/1078-0432.CCR-15-2971.
Ferris RL, Lenz HJ, Trotta AM, García-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. 2018;63:48–60. https://doi.org/10.1016/j.ctrv.2017.11.008.
Boland PM, Hutson A, Maguire O, Minderman H, Fountzilas C, Iyer RV. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. J Clin Oncol. 2018;36:834. https://doi.org/10.1200/jco.2018.36.4_suppl.834.
Stein A, Binder M, Goekkurt E, Lorenzen S, Riera-Knorrenschild J, Depenbusch R, et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): Final results of the phase II AVETUX trial (AIO-KRK-0216). J Clin Oncol. 2020;38:96. https://doi.org/10.1200/jco.2020.38.4_suppl.96.
Lee MS, Loehrer PJ, Imanirad I, Cohen S, Ciombor KK, Moore DT, et al. Phase II study of ipilimumab, nivolumab, and panitumumab in patients with KRAS / NRAS / BRAF wild-type (WT) microsatellite stable (MSS) metastatic colorectal cancer (mCRC). J Clin Oncol. 2021;39:7. https://doi.org/10.1200/jco.2021.39.3_suppl.7.
Liu P, Wang Y, Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B. 2019;9:871–9. https://doi.org/10.1016/j.apsb.2019.03.002.
Taieb J, Lapeyre-Prost A, Laurent Puig P, Zaanan A. Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br J Cancer. 2019;121:434–42. https://doi.org/10.1038/s41416-019-0526-2.
• Carvalho PD, Guimarães CF, Cardoso AP, Mendonça S, Costa ÂM, Oliveira MJ, et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. 2018. https://doi.org/10.1158/0008-5472.CAN-17-2084 These reference are of importance because clearly takes stock of the impact of Kras mutations in cancer cells on tumor immunogenicity and immune cells phenotype and activity inside TME.
Petanidis S, Anestakis D, Argyraki M, Hadzopoulou-Cladaras M, Salifoglou A. Differential expression of IL-17, 22 and 23 in the progression of colorectal cancer in patients with K-ras mutation: Ras signal inhibition and crosstalk with GM-CSF and IFN-γ. PLoS One. 2013;8:e73616. https://doi.org/10.1371/journal.pone.0073616.
Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44:609–21. https://doi.org/10.1016/j.immuni.2016.01.024.
Mimura K, Shiraishi K, Mueller A, Izawa S, Kua L-F, So J, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol. 2013;191:6261–72. https://doi.org/10.4049/jimmunol.1301597.
Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499–509. https://doi.org/10.1158/1078-0432.CCR-15-1125.
Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol Res. 2014;2:351–60. https://doi.org/10.1158/2326-6066.CIR-13-0181.
•• Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019. https://doi.org/10.1038/s41586-019-1694-1 These references are of outstanding importance because analyzed the activity of the KRAS G12C inhibitor, paving the way to the possibility of hitting a target untargettable until now, and showing its immunomodulatory properties and, therefore, sinergism with immune check-point inhibitors.
•• Fakih M, Desai J, Kuboki Y, Strickler JH, Price TJ, Durm GA, et al. CodeBreak 100 Activity of AMG 510, a novel small molecule inhibitor of KRAS G12C in patients with advanced colorectal cancer. J Clin Oncol. 2020. https://doi.org/10.1200/jco.2020.38.15_suppl.4018 These references are of outstanding importance because analyzed the activity of the KRAS G12C inhibitor, paving the way to the possibility of hitting a target untargettable until now, and showing its immunomodulatory properties and, therefore, sinergism with immune check-point inhibitors.
Johnson ML, Ou SHI, Barve M, Rybkin II, Papadopoulos KP, Leal TA, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur J Cancer. 2020;138:S2. https://doi.org/10.1016/S0959-8049(20)31077-7.
Fakih M, Durm GA, Govindan R, Falchook GS, Soman N, Henary HA, et al. Trial in progress: a phase Ib study of AMG 510, a specific and irreversible KRAS G12C inhibitor, in combination with other anticancer therapies in patients with advanced solid tumors harboring KRAS p.G12C mutation (CodeBreak 101). J Clin Oncol. 2020. https://doi.org/10.1200/jco.2020.38.15_suppl.tps3661.
Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20:849–61. https://doi.org/10.1016/S1470-2045(19)30027-0.
Gordon S, Plüddemann A. The mononuclear phagocytic system. Generation of diversity Front Immunol. 2019;10. https://doi.org/10.3389/fimmu.2019.01893.
Anfray C, Ummarino A, Andón FT, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. 2019;9. https://doi.org/10.3390/cells9010046.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. https://doi.org/10.1038/nrclinonc.2016.217.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9. https://doi.org/10.1038/nature22396.
Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18:60. https://doi.org/10.1186/s12943-019-0974-6.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38:2053–61. https://doi.org/10.1200/JCO.19.03296.
Cousin S, Cantarel C, Guégan J-P, Gomez-Roca C, Metges J-P, Adenis A, et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (REGOMUNE) : a single arm, open-label, phase II trial. Clin Cancer Res. 2021. https://doi.org/10.1158/1078-0432.ccr-20-3416.
Iwasa S, Okita N, Kuchiba A, Ogawa G, Kawasaki M, Nakamura K, et al. Phase II study of lenvatinib for metastatic colorectal cancer refractory to standard chemotherapy: the LEMON study (NCCH1503). ESMO Open. 2020;5:e000776. https://doi.org/10.1136/esmoopen-2020-000776.
Funding
Open access funding provided by Università Politecnica delle Marche within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Federica Pecci declares that she has no conflict of interest. Luca Cantini declares that he has no conflict of interest. Alessandro Bittoni declares that he has no conflict of interest. Edoardo Lenci declares that he has no conflict of interest. Alessio Lupi declares that he has no conflict of interest. Sonia Crocetti declares that she has no conflict of interest. Enrica Giglio declares that she has no conflict of interest. Riccardo Giampieri declares that he has no conflict of interest. Rossana Berardi has served as a consultant/advisory board member for AstraZeneca, Boehringer Ingelheim, Novartis, MSD, Otsuka Pharmaceutical, Eli Lilly, and Roche.
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lower Gastrointestinal Cancers
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pecci, F., Cantini, L., Bittoni, A. et al. Beyond Microsatellite Instability: Evolving Strategies Integrating Immunotherapy for Microsatellite Stable Colorectal Cancer. Curr. Treat. Options in Oncol. 22, 69 (2021). https://doi.org/10.1007/s11864-021-00870-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s11864-021-00870-z