
Abstract
As an essential regulatory component in plants, microRNAs (miRNAs) have been intensively studied over the past decade. Although hundreds of miRNAs have been identified and analyzed in many important crops and model plants, very little is known about the function of common wheat (Triticum aestivum L.) miRNAs. In this study, we performed computational prediction of novel wheat miRNAs based on BLAST searches of the expressed sequence tag database. The expression profiles of all miRNAs were performed for both vegetative and reproductive tissues to identify developmentally regulated miRNAs. A total of 19 new miRNAs belonging to 12 MIR families were identified using stringent criteria for miRNA annotation. For all of the miRNAs, the secondary structures of their precursor sequences were predicted. Two pairs of distinct miRNAs were found to be located on the same precursor. The predicted miRNAs were experimentally verified by a stem-loop qRT-PCR-based assay. The expression profiles were performed in both vegetative and reproductive tissues to find the potential correlations between the developmental phase and miRNA activity. Thirteen out of 19 miRNAs were upregulated at certain phases of plant development, and three of them (miR319, miR395, and miR171) showed the greatest expression in young spikes during microsporogenesis. Our results provide useful information for future studies of miRNA-mediated regulation of flower and grain development in wheat.
Similar content being viewed by others


Introduction
For almost two decades, small RNAs have become one of the major research topics in both animals and plants. MicroRNAs (miRNAs) are a class of small, 19–24 nt-long non-coding RNA species originating from double-stranded miRNA precursors. The discovery of miRNA (MIR) genes and their function has revolutionized the understanding of gene expression regulation in eukaryotic organisms. In plants, MIR genes are found mainly in intergenic regions and are transcribed by RNA polymerase II to primary transcripts (pri-miRNAs), which form an imperfect fold-back structure. This structure is further processed to a stem-loop precursor from which mature duplex miRNA is excised by a DLC-like protein (Voinnet 2009). The mechanism of gene silencing by miRNAs is analogous to a siRNA pathway. The majority of plant miRNAs guide the RNA-induced silencing complex (RISC complex) to the target mRNA which is subsequently degraded (Bartel 2009); however, mRNA translation inhibition and direct DNA methylation were also reported (Brodersen et al. 2008; Lanet et al. 2009; Wu et al. 2010; Yu and Wang 2010).
In plants, MIR genes account for a diverse group in terms of structure, genome localization, and expression patterns. The size of known Arabidopsis MIR genes ranges from about 200 to over 3000 bp, and size differences may occur in the same family of genes encoding almost identical miRNAs. MIR genes may contain single or multiple introns and their transcripts can be processed by alternative splicing. Most of the MIR genes form independent transcriptional units. Moreover, some of them are also localized in introns or UTR regions of protein coding genes, and some partially overlap with the coding sequence of other genes. Clusters of MIR genes and polycistronic miRNAs have also been reported (Baldrich et al. 2016). The above examples show how complicated the processes are with respect to the expression regulation of both miRNAs and their target genes.
Plant miRNAs are engaged in the regulation of key developmental processes such as leaf development (Palatnik et al. 2003; Laufs et al. 2004; Schwab et al. 2005), leaf polarity (Vaucheret et al. 2004; Williams et al. 2005), floral organ identity, or flowering time (Aukerman and Sakai 2003; Chen 2004; Mallory et al. 2004, 2005). Moreover, miRNAs are involved in phytohormone signaling pathways by the regulation of their metabolism, distribution, and perception (Curaba et al. 2014). The second major group of processes regulated by miRNAs is adaptive responses to abiotic and biotic stress (reviewed by Khraiwesh et al. 2012).
In recent years, the methodology of the identification and characterization of miRNAs has been greatly improved for both experimental and bioinformatics approaches. Apart from conventional experimental techniques such as cloning, RNA gel blot, and in situ hybridization, the high-throughput methods including NGS (next-generation sequencing) and microarray technology have also been applied for the identification and activity screening of miRNAs. Deep sequencing of small RNA libraries is very effective for the identification of new miRNAs, especially those activated by stress responses. It is possible to distinguish members of miRNA families at a single nucleotide resolution which is difficult with other methods. Deep sequencing is often performed in combination with parallel analysis of RNA ends analysis (PARE), a modified 5′ RACE technique allowing for mRNA cleavage site mapping and for the validation of miRNA-target genes (German et al. 2008). PARE analysis can be used for the identification and validation of miRNA-target RNA pairs at a massive scale (German et al. 2008; Jeong et al. 2011, 2013). However, these approaches generate enormous sets of data which require intensive bioinformatical processing to be correctly interpreted. Much caution should be taken while analyzing the results of PARE and deep sequencing, since distinguishing miRNAs from siRNAs (which closely resemble mature miRNAs in their structural and biochemical characteristics) and from mRNA decay products is particularly challenging. Moreover, the relatively high cost of NGS and microarrays is a limiting factor for the routine application of these methods. The bioinformatics approach can be to some degree a fast and cheap alternative for experimental methods. The computational prediction of miRNAs relies on specific characteristics of both mature miRNA and its precursor sequences. Based on these characteristics, the secondary structure of miRNA precursors can be predicted using available tools such as the mfold web server (Zuker 2003). Other bioinformatics tools, e.g., psRNATarget (Dai and Zhao 2011) or TAPIR (Bonnet et al. 2010) have also been developed for the prediction of miRNA-target genes, however, the accuracy of these predictions is limited in the case of some organisms by the number of available annotated genomic sequences. A commonly used strategy for the identification of new miRNAs is BLAST searching of the GSS or expressed sequence tag (EST) databases to find conserved homologs of already known miRNAs. The hit sequences are analyzed and filtered to select those with potential hairpin structure and to remove protein coding sequences. The putative miRNA sequences are then validated based on criteria proposed for miRNA annotation. Although only conserved miRNAs can be found by blast searches, the vast amount of plant miRNAs have been identified by this strategy (Yao et al. 2007; Dryanova et al. 2008; Sunkar and Jagadeeswaran 2008; Zhou et al. 2008).
Despite the economic importance of common wheat (Triticum aestivum), which is the third world’s major cereal crop after rice and maize, the knowledge of wheat microRNAs is far less advanced when compared to the top two crops and model plants. At the time of this study, only 119 wheat miRNAs were registered in miRBase (release 21), while the number of registered Brachypodium, rice, and maize miRNAs was 525, 713, and 321, respectively. The size (17 Gb) and complexity of the hexaploid wheat genome are a major limitation in miRNA identification and activity screening. The identification of target genes and the functional analysis of wheat miRNAs are also difficult, since the wheat genome has not been assembled and is poorly annotated. Recently, numerous miRNAs have been identified in wheat by deep sequencing of small RNA libraries (Yao et al. 2007; Wei et al. 2009; Xin et al. 2010; Kantar et al. 2012; Meng et al. 2013; Sun et al. 2014; Akdogan et al. 2016), however, most of them have not been registered in the available databases. It has been observed that many wheat miRNAs are regulated by stress conditions such as drought (Akdogan et al. 2016) or fungal infections (Xin et al. 2010). Some of them are also associated with morphology and developmental processes including plant height and flowering (Kantar et al. 2012) or grain development (Meng et al. 2013; Sun et al. 2014).
The knowledge about biological function of wheat miRNAs, their regulation and tissue specificity is also important in functional genomics studies with the use of artificial microRNA (amiRNA). amiRNA-based gene silencing is a powerful technology which allows for highly efficient and precise gene knock-out and was successfully applied to wheat (Fahim et al. 2012; Gasparis et al. 2017). However, this technique requires the use of a valid, functional miRNA precursor which is recognized and processed by cellular RNAi machinery.
Here, we present a bioinformatics approach for the identification of novel conserved wheat miRNAs in combination with fast, sensitive, and cost-effective experimental validation. For miRNA detection and expression profiling we adopted the previously developed stem-loop qRT-PCR assay (Varkonyi-Gasic et al. 2007) with the use of a LNA-based universal probe. This is a very sensitive method allowing for miRNA detection from as little as 20 pg of total RNA; thus, even very low expressing miRNAs can be detected. The application of a universal probe (one for all miRNAs) significantly reduces the cost of the experiment. The expression profiles of all miRNAs were performed for both vegetative and reproductive tissues to identify developmentally regulated miRNAs. The possible correlation between miRNA expression and plant development stages are discussed.
Materials and methods
BLAST search of homologue miRNA sequences
Potential wheat miRNAs were identified based on their homology with mature miRNA sequences of Arabidopsis thaliana, Brachypodium dystachion, Zea mays, and Oryza sativa. The sequences of previously known miRNAs of these species were obtained from miRBase (Griffiths-Jones 2004, 2010; Griffiths-Jones et al. 2006; Kozomara and Griffiths-Jones 2011, 2014) excluding homologues of previously identified wheat miRNAs. The miRNA sequences were then subjected to a BLASTN search against T. aestivum ESTs in the NCBI database with the following parameters: expected threshold was set to 1000, the number of aligned sequences was raised to 1000, and the word size matching the query and database sequences was set to 7.
All BLAST results were saved in Excel spreadsheets. Only closely matching EST sequences with a maximum of three mismatches (n − 3, where n is the length of the query miRNA) were selected for further analysis. The selected ESTs were used for BLASTX analysis. The protein coding sequences were removed and non-coding sequences were used for the prediction of secondary hairpin stem-loop structures of potential miRNA precursors.
Prediction of the secondary structures of miRNA precursors
The secondary RNA structures of selected EST sequences were generated using the Zuker folding algorithm (Zuker 2003) with the mfold web server (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form). EST sequences extending from 300 nt upstream to 300 nt downstream of the blast hit or shorter were used for the hairpin structures prediction with the following default parameters: folding temperature was fixed at 37 °C, ionic conditions were set at 1 M NaCl without divalent ions. Structures with the highest minimal free energy were selected and the MFEI index (minimal folding free energy index) were designated as described by Zhang et al. (2006b). The potential miRNA precursors were designated as described by Meyers et al. (2008). The RNA sequences were considered miRNA candidates only if they fulfilled the following criteria: (1) an RNA sequence was able to fold into an appropriate stem-loop hairpin structure; (2) a mature miRNA sequence was located in one arm of the hairpin structure; (3) miRNAs contained less than six mismatches with the opposite miRNA* sequence on the other arm; (4) no loops or breaks in the miRNA* sequences; (5) the predicted secondary structures had greater MFEIs and negative MFEs; (6) the predicted mature miRNAs had no more than four mismatches as compared with the corresponding homologue mature miRNA. The predicted structures were also validated on Wheat MicroRNA Portal (http://wheat.bioinfo.uqam.ca) (Agharbaoui et al. 2015; Remita et al. 2016) using mirDup (v1.2) (Leclercq et al. 2013) with two datasets: all miRBase (B) r20 and Wheat (W) r20.
Prediction of the miRNA-target genes
Mature sequences of each predicted miRNA were used to find potential target genes in wheat using the online software psRNATarget (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao 2011). The searches were done in T. aestivum unigene, DFCI Gene Index version 12 with the default parameters, except the maximum expectation which was set to 5. Potential target genes were also searched on Wheat MicroRNA Portal using TAPIR web server (Bonnet et al. 2010).
Plant material
The Chinese Spring cultivar of wheat (T. aestivum L.) was used in the experiments. The plants were grown under controlled conditions, 18/15 °C day/night with a 16-h photoperiod as described by Gasparis et al. (2013).
Detection and quantification of predicted miRNAs
Total RNA was isolated using TRI Reagent (Sigma-Aldrich) following manufacturer’s instructions. To remove contaminating gDNA, the samples were treated with DNase I (Thermo Scientific) according to manufacturer’s protocol. Low molecular weight RNA containing the microRNA fraction was separated from the total RNA using polyethylene glycol (PEG) as described by Goto et al. (Goto et al. 2003). 250 ng of RNA from each sample was used for the reverse transcription reaction with Maxima reverse transcriptase (Thermo Scientific) and 0.05 µM of stem-loop RT primer (Additional file 1: Table S1). The primers were designed according to Varkonyi-Gasic et al. (Varkonyi-Gasic et al. 2007). The loop region of the RT primers contained a sequence complementary to the UPL probe #21 (Roche). The samples were first denatured at 65 °C for 5 min and chilled on ice. Afterward, the reverse transcription reaction was performed using the following program: 94 °C for 2 min followed by 40 cycles of 94 °C for 15 s and 60 °C for 1 min. The reaction was terminated at 70 °C for 10 min. No-RT primer and no-RNA reactions were included as negative controls, and a sample with RT primer for tae-miR164 was used as a positive control for all of the reactions. The qPCR reaction was carried out in a 20 µl reaction mixture containing 10 µl of TaqMan Fast Advanced Master Mix (Applied Biosystems), 2 µl of cDNA sample, 0.4 μM of each primer and 0.25 µl of UPL probe #21. The miRNA specific forward primers and universal reverse primer (Varkonyi-Gasic et al. 2007) and U6 RNA primers are listed in Table S1 (Supplementary Table 1). The reaction was run in a RotorGene Q thermal cycler (Qiagen) using the following program: initial denaturation step at 95 °C for 2 min followed by 55 cycles of amplification at 95 °C for 5 s, 60 °C for 15 s, and 72 °C for 5 s. U6 RNA was used as a reference gene. A series of dilutions of the amplified miRNAs and the U6 RNA were used in the first qPCR reaction to estimate the reaction efficiency. All reactions were performed in triple replications. The relative level of miRNAs was quantified using the Pfaffl method (Pfaffl 2001). The expression values were median centered by arrays and genes, and hierarchically clustered (average linkage correlation metric) using the Cluster program from Stanford University (de Hoon et al. 2004). The heatmap was generated in the Treeview program (Saldanha 2004). Expression that had a value equal to 1 was designated in black, greater expression was designated in yellow, and lower expression was designated in blue.
Results
Conservation of wheat miRNAs
For BLAST searches, we chose two model plants, A. thaliana and B. dystachion representing dicots and monocots, respectively, and two important cereal crops, rice (O. sativa) and maize (Z. mays) which simultaneously have a large number of identified miRNAs.
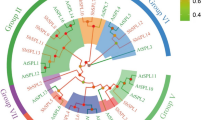
Before performing the BLAST analysis, we searched the miRBase database to select all of the known miRNAs conserved between these species and wheat (Fig. 1). Among 54 MIR families found in query searches, 29 are conserved within both dicots and monocots, including 17 families containing wheat miRNAs. The remaining 25 are specific to monocots only, including 14 families containing wheat miRNAs. All miRNAs homologous to known wheat miRNAs were excluded from the BLAST analysis to reduce the number of outcome sequences and to eliminate false positives.

Conservation of microRNA families between species used in this study. The miRNAs which are conserved between dicots and monocots are marked green, and blue represents only the monocot-specific miRNAs. The dark green and dark blue colors represent the families of which new miRNAs were identified in this study
Identification of new miRNA candidates
To identify new wheat MIR candidates, we used mature miRNA sequences of Arabidopsis, Brachypodium, rice, and maize, for BLASTN analysis against the whole EST database of wheat (1,298,692 sequences). The BLASTN searches were done separately for each species and the number of miRNAs used was 337 for Arabidopsis, 454 for Brachypodium, 56 for rice, and 187 for maize. EST sequences (458,318) found in the BLAST searches were further analyzed and filtered as shown in Fig. 2. The main criteria utilized at the first step of selection of the MIR candidates were sequence similarity and the ability to form hairpin structures. ESTs with a maximum of 3 mismatches in the mature miRNA sequences which were able to form potential hairpin structures and did not encode proteins were chosen for further selection. To exclude ESTs created from the same RNA, ESTs containing the same length hairpin sequence were blasted against each other. ESTs with the highest similarity (E value <e−100) were considered as the same miRNA precursor; thus, only ESTs with the longest sequences flanking the mature miRNA were chosen for secondary structure prediction. As a result, 19 new miRNA candidates were identified belonging to 12 MIR families (Table 1). The sequences were homologous to Brachypodium, maize, and rice miRNAs, and no homologs of Arabidopsis miRNAs were found. Except for two candidates (miR319 and miR9493), all of the miRNAs were 21 nt long, which is the most typical length of a mature miRNA. All of the candidates start with the same base as their homolog, and 12 out of 19 miRNAs start with a 5′ uridine, a very common feature for known miRNAs.

Schematic representation of the procedure used for the identification of homologous wheat miRNAs
The putative miRNAs were blasted in miRBase against all monocotyledon classes and the most similar sequences were aligned (see Additional file 1: Fig. S1). This allowed us to find the closest homolog for each miRNA candidate and to assign it to the appropriate MIR family. MIR5180, MIR444, and MIR5181 were the most represented families containing, respectively, 4, 3, and 3 miRNA candidates. For the rest of the MIR families, only one candidate was found. Most of the miRNA candidates show high sequence similarity to their homologs, containing only 1 or 2 mismatches, and six miRNAs (miR169, miR171, miR395, miR444.1, miR444.2, and miR444.3) are identical with the corresponding homologs. As expected, the highest sequence similarity is observed in the highly conserved MIR families specific to both dicots and monocots such as MIR169, MIR171, and MIR395. All miRNA candidates assigned to these families are identical with at least one member. The MIR319 family is here an exception, as a miR319 candidate is closely similar (with 3 mismatches) to only one miRNA; however, this family is much smaller than mentioned above. MIR444 is a highly conserved, monocot-specific family where miRNAs from different species are often identical. All three miRNA candidates assigned to this family are also identical with Brachypodium and barley miRNAs. In contrast, the miRNA candidates assigned to less conserved families such as MIR5067, MIR5175, MIR5180, MIR5181, MIR7742, and MIR9493 are more distant from other members (Supplementary Fig. 1). According to our knowledge, the MIR7742 and MIR9493 families were identified for the first time in wheat, while the other 17 miRNAs are new, consecutive members of already existing wheat MIR families (Table 1).
Prediction of precursor secondary structures and designation of new wheat miRNAs
The secondary structure of the precursors of the identified miRNAs was predicted using the mfold web server (Supplementary Fig. 2). The structures with the greatest minimal free energy which simultaneously do not have side chains in the “stem” region, have no more than six mismatches between the miRNA and miRNA* sequence are considered as precursor miRNAs. Additionally, the minimal free energy index (MFEI) was calculated for each precursor. The MFEI values ranged from 0.79 to 1.85 (Table 2). These results are in agreement with the results of Zhang et al. (Zhang et al. 2006b) who observed that the MFEI is significantly greater in miRNA than other non-coding RNAs. To designate new miRNAs, we adopted the criteria proposed previously (Zhang et al. 2006a; Meyers et al. 2008). Using these selection criteria, we designated 19 new wheat miRNAs created from 16 precursors (Table 2).
Interestingly, two pairs of miRNA are located on the same precursor. miR5180.1 and miR5181.1 are located on the 5′ arm of the precursor identified in EST HX178752.1. The sequences of both of the miRNAs are slightly similar and they partially overlap (Fig. 3a). The second pair is two distinct miRNAs, miR319 and miR7742, which were found on a precursor encoded by EST HX145825.1; however, miR319 is located on the 5′ arm and miR7742 on the 3′ arm of the precursor (Fig. 3b).

Examples of polycistronic wheat miRNA precursors containing two non-homologous miRNAs identified in ESTs HX178752.1 (a) and HX145825.1 (b)
All mature miRNAs are found on the same arm of the precursor (either 5′ or 3′) as their previously known homologs which confirm that they originate from the same progenitor.
Moreover, the complementarity between the miRNAs and their corresponding miRNAs* meets the requirements for miRNA genes, as there are no more than four mismatches in all of the precursors. The predicted miRNAs were also validated by mirDup (v1.2) software. This tool uses an algorithm trained with experimentally validated miRNAs from miRbase to compare candidate structures with known miRNAs. As shown in Supplementary Table 2 all 19 miRNA candidates were positively validated using either all miRBase (B) or Wheat (W) datasets. In case of three miRNAs, there were contradictory results between these datasets.
Potential targets of identified miRNAs
The knowledge of miRNA-target genes is essential in determining their function.
Potential targets of new miRNAs in wheat were predicted using the psRNATarget web server (Table 3). The targets of eight miRNAs were predicted with a stringent cut-off threshold (maximum expectation from 0 to 2) which reduces the false positive predictions. The prediction of targets for the remaining eleven miRNAs was possible only with a more relaxed cut-off threshold (maximum expectation from 3 to 5). The predicted targets are different types of transcription factors and functional proteins inhibited either by cleavage or translational repression. The target genes of the miR169 and miR444 families, the CAAT-box transcription factor, and the MIKC-type MADS-box transcription factor, respectively, have also been reported as target genes of maize and rice miRNAs (Yan et al. 2014; Luan et al. 2015). The remaining 15 target genes are unique for wheat only. Among them, the same targets for miR5067, miR5180.2, and miR5181.3 as predicted here were also reported by other researchers (Kantar et al. 2012).
Other target genes were found on Wheat MicroRNA Portal but only for four miRNAs (Table 4). These are cDNA clones derived from transcripts isolated mainly from roots and meiotic anthers. The useful information available through this search tool is the number of known wheat miRNAs associated with predicted target genes. Moreover, these associated miRNAs are assigned to sequenced wheat libraries generated under different experimental conditions (see description below the Table 4) and their expression regulation is also provided. Wheat miRNAs associated with the predicted target genes belong to the same MIR families as miRNAs predicted in this study (except miR7742) and are upregulated by different conditions such as vernalisation, cold tolerance and salt response (miR444) or aluminum response (miR319, miR159).
Detection of new wheat miRNAs and their expression profiles
To detect transcripts of the identified miRNAs, we performed a stem-loop qRT-PCR assay. Using this method, we detected all 19 miRNAs in both vegetative and reproductive tissues including leaves, roots, and spikes at different developmental stages. Next, the relative expression of each miRNA was quantified in qPCR reactions using the universal probe UPL#21 and U6 RNA for normalization. The expression profiles were designated for each miRNA in 7-day-old seedling leaves and roots, 8-week-old leaves, young spikes at the microsporogenesis stage, and spikes at 0 DAP and 14 DAP (days after pollination). Since the expression level in 8-week-old leaves was most aligned among all of the miRNAs, this sample was used as a calibrator with the expression set to 1. The miRNA expression in other tissues was calculated as x-fold increase or decrease against the calibrator. To better track the changes in expression, the data for all of the miRNAs were clustered and converted into a heat map (Fig. 4). Because of technical limitations, the color bars do not reflect the real differences in expression, therefore the fold change values are presented in Supplementary Fig. 3. As shown in Fig. 4, several miRNAs were upregulated at certain developmental stages, whereas the expression of the others remained unchanged. The group of 11 miRNAs was highly upregulated in young spikes with the highest expression at the microsporogenesis stage which slightly decreased at 0 DAP. Within this group the cluster of three miRNAs (miR319, miR395, and miR171) showed the highest upregulation during microsporogenesis, where the expression of miR319 was extremely high (2256 times higher than in 8-week-old leaves). The expression of all of the miRNAs in other tissues was relatively lower and stable, except for miR169 which was highly expressed in seedling leaves and miR444.3 which showed greater expression in seedling roots. The lowest expression level of all of the identified miRNAs was detected in 14 DAP spikes. Interestingly, we observed differences in the expression pattern among members of the same MIR family. For example, miR444.3 showed greater expression in roots, in contrast to two other miRNAs, miR444.1 and miR444.2. Similarly, the expression of miR5180.1 in spikes at microsporogenesis was approximately 2 times greater when compared with three other members of this family.

Expression profiles of newly identified wheat miRNAs from different tissues; (A) young spikes at the microsporogenesis phase, (B) 0 DAP spikes, (C) 14 DAP spikes, (D) 7-day-old seedling leaves, (E) 8-week-old leaves, (F) 7-day-old seedling roots. Yellow denotes greater expression, blue denotes lower expression, and black no changes in expression. The range of the expression values is from −3.74 to +3.74 fold; however, both the maximum and minimum level of expression may exceed these values
Discussion
Monocot-specific miRNAs are more conserved between closely related species
Computational prediction of novel miRNAs relies on the sequence homology of conserved mature miRNA sequences and specific features of their double-stranded precursors. Numerous MIR genes are evolutionarily conserved throughout the plant and animal kingdoms. In the plant kingdom, several MIR families such as MIR156, MIR160, MIR166, MIR171, MIR319, and MIR395 have been identified in all major lineages including mosses, gymnosperms, monocots, and dicots. The strong conservation of some plant miRNAs allows for sequence analysis using whole genome sequences or ESTs even between unrelated species. In our study, we used an EST database for blast analysis as a large source (several times larger than a GSS database) of new potential miRNAs that are expressed in wheat. Therefore, one should expect that these miRNAs should be functional. This approach was successfully used for the identification of new miRNAs in plants (Zhang et al. 2005; Yao et al. 2007; Dryanova et al. 2008; Sunkar and Jagadeeswaran 2008; Zhou et al. 2008; Unver and Budak 2009).
miRNA sequences of main monocot species with the greatest number of registered miRNAs were used in blast analysis. 525 miRNAs of Brachypodium, 713 of rice, and 321 of maize have been registered at miRBase (release 21). In comparison, the number of wheat miRNAs is only 119. It is worth noting that the major portion of the rice and Brachypodium miRNAs, the model plants of monocot crops, have been identified and experimentally validated by cloning and sequencing (Zhang et al. 2009; Jeong et al. 2011; Wei et al. 2011; Jeong et al. 2013), therefore these miRNAs are a reliable source for the search of conserved homologues in other plants. Despite a high degree of conservation of numerous MIR genes, the majority of plant miRNAs is conserved only among families or is unique for particular species (Cuperus et al. 2011). As expected, most of the miRNAs predicted by us (15 out of 19) are monocot specific. Among them, 11 miRNAs were common only for Brachypodium and wheat, which are more closely related with themselves than with rice and maize. As shown in Fig. 1, among monocot-specific MIR families 19 are common for Brachypodium and wheat, compared to only 6 families for rice and maize. Moreover, the monocot-specific miRNAs exhibit greater sequence diversity within the MIR families than the more conserved miRNAs present in both monocot and dicot plants (see Supplementary Fig. 1). The above observations may suggest that monocot-specific miRNAs as evolutionarily younger have emerged as a result of adaptive processes and undergone more dynamic variation. This also implies that these miRNAs are more connected with stress response processes rather than with developmental regulation, which is controlled by more conserved miRNAs.
To identify new wheat miRNAs and their precursors, we applied the previously proposed and accepted criteria of annotation (Zhang et al. 2006a; Meyers et al. 2008). The secondary structure of a miRNA precursor is usually predicted based on sequences flanking the 300 nt upstream and 300 nt downstream of the mature miRNA, as typical precursors rarely exceed this size. Some of the EST sequences selected by us were shorter; however, all of the predicted structures met the requirements specified for miRNA precursors. An interesting case is two precursors found on the HX178752.1 and HX145825.1 ESTs, each including two pairs of distinct miRNAs. The first pair is located on the same arm of the precursor and they partially overlap, in turn miRNAs of the second pair are located on the opposite arms of the precursor. Polycistronic MIR genes in which multiple miRNAs are located within a single transcriptional unit have already been reported in rice (Baldrich et al. 2016). Both homologous (e.g., miR169m, miR169l, and miR169q) and non-homologous (e.g., miR14233 and miR1868) polycistronic miRNAs have been observed.
Highly conserved wheat miRNAs are developmentally regulated
Expression profiles were performed for all of the identified miRNAs in both vegetative and generative tissues at important developmental stages. By the UPL probe containing synthetic LNA nucleotides with increased affinity to the complementary strand, the sensitivity of the assay is enough to detect miRNA from as little as 20 pg of total RNA isolated from plant tissue, thus it is a more applicable method to quantify even very low levels of miRNA than Northern blot.
The tested plants were grown in typical conditions for wheat since we intended to perform expression profiling of miRNAs during normal growth conditions without influence of any stress factors. As shown, the tested miRNAs can be divided into three main groups based on expression profiles. A group of eleven miRNAs highly upregulated in young spikes, a group of three miRNAs upregulated in seedling roots (miR444.3) and leaves (miR169 and miR164), and a group of six miRNAs which did not indicate any significant changes in expression. miR164 was previously cloned in wheat (Yao et al. 2007) and was used here as a positive control for all of the reactions. From the group of miRNAs upregulated in young spikes stands out a cluster of three miRNAs (miR319, miR395, and miR171) with particularly high expression at the microsporogenesis stage. Their expression is dozens of times greater, and in the case of miR319 above two thousand times greater than in leaves. Such extremely high levels of expression may suggest an important role for miR319 at this phase of inflorescence development. MIR319 is a strongly conserved family in higher plants. In dicotyledonous plants, miR319 and its target genes, including TCP transcription factors affect leaf morphogenesis (Palatnik et al. 2003; Koyama et al. 2007; Ori et al. 2007; Koyama et al. 2010) and flower development (Nag et al. 2009). In rice and wheat, miR319 targets the MYB transcription factor and its expression can be induced under cold and drought stress conditions. miR319 and miR395 are strongly induced in leaves of wheat and rice during exposure to drought stress conditions, however, their activity in flower tissues was not tested (Zhou et al. 2010; Yang et al. 2013; Akdogan et al. 2016). In cereals, the reproductive phase and particularly the meiosis stage is highly vulnerable to drought stress, which can delay or completely inhibit flowering or cause pollen sterility (Saini and Westgate 1999). Perhaps the upregulation of these miRNAs at the reproductive phase is a natural mechanism protecting against drought effects.
Tandem miR169 and its target gene, the CAAT-box transcription factor complex play critical roles in plant development, which is confirmed by the high expression of miR169 in seedling leaves. However, there are several reports indicating that miRNAs from the MIR169 family are also involved in response to biotic and abiotic stresses (Sunkar et al. 2012; Inal et al. 2014; Luan et al. 2015). Interestingly, under drought stress the wheat miR169 was highly downregulated in leaves but upregulated in roots (Akdogan et al. 2016). In turn, the greater expression of miR444.3 in roots is in agreement with the observations in rice in which miR444 regulate the \({\text{NO}}_{3}^{ - }\) signaling pathway and root development. MIKC-type MADS-box transcription factors, the target genes of miR444 predicted in this study are orthologues of similar genes in rice (Yan et al. 2014). It is also possible that miR444 regulates the expression of other genes in roots in response of cold treatment. Two target genes of miR444.3 listed in Table 4 (CK200584.1 and BE405735.1) are transcripts isolated from cold-acclimated roots and other wheat miRNAs associated with these genes are upregulated under cold tolerance conditions.
The expression profiles of the remaining monocot-specific miRNAs are less unambiguous. Some of them were slightly upregulated in young spikes, while others did not show any significant expression changes, which may suggest that they are induced by environmental stresses. Unfortunately, very little is known about these miRNAs in other species and no similar wheat miRNAs were found in Wheat MicroRNA Portal to make any conclusions about their possible functions.
Conclusion
All of the tested miRNAs are constitutively expressed at a minimum level allowing their detection, and some of them are strongly upregulated at certain developmental stages, while the remaining are probably induced in response to stress factors. Moreover, the expression levels of four highly conserved miRNAs (i.e., miR169, miR171, miR319, and miR395) were greater than the monocot-specific miRNAs. This result is in agreement with the previous observations which indicate that highly conserved miRNAs common to higher plants are expressed at greater levels than less conserved or species-specific miRNAs (Willmann and Poethig 2007; Cuperus et al. 2011).
Although numerous wheat miRNAs have been identified recently, the knowledge about their functions is far less advanced compared to other important monocot crops. Most of the wheat miRNA-target genes were predicted in silico and only a small fraction were experimentally validated. The size and complexity of the hexaploid wheat genome is a major limiting factor for the functional analysis of miRNAs. Studies of other plants revealed that numerous miRNAs are involved in adaptive responses to abiotic stresses such as cold and drought tolerance. The similar findings in wheat may have important implications for possible breeding applications. While the number of identified miRNAs in model plants is constantly increasing, the computational prediction combined with highly sensitive detection methods is a cost-effective and efficient alternative for the identification of novel conserved miRNAs in wheat.
Author contribution statement
SG designed the experiment, performed part of experimental work and bioinformatics analyses, and wrote the manuscript. YY performed part of experimental work and bioinformatics analyses. AN-O designed the experiment, guided all aspects of the research project and revised the manuscript. All authors read and approved the final manuscript.
References
Agharbaoui Z, Leclercq M, Remita MA, Badawi MA, Lord E, Houde M, Danyluk J, Diallo AB, Sarhan F (2015) An integrative approach to identify hexaploid wheat miRNAome associated with development and tolerance to abiotic stress. BMC Genom 16:339
Akdogan G, Tufekci ED, Uranbey S, Unver T (2016) miRNA-based drought regulation in wheat. Funct Integr Genom 16:221–233
Aukerman MJ, Sakai H (2003) Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15:2730–2741
Baldrich P, Hsing YI, San Segundo B (2016) Genome-wide analysis of polycistronic microRNAs in cultivated and wild rice. Genome Biol Evol 8:1104–1114
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Bonnet E, He Y, Billiau K, Van de Peer Y (2010) TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26:1566–1568
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Chen X (2004) A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 303:2022–2025
Cuperus JT, Fahlgren N, Carrington JC (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442
Curaba J, Singh MB, Bhalla PL (2014) miRNAs in the crosstalk between phytohormone signalling pathways. J Exp Bot 65:1425–1438
Dai XB, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
de Hoon MJ, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454
Dryanova A, Zakharov A, Gulick PJ (2008) Data mining for miRNAs and their targets in the Triticeae. Genome 51:433–443
Fahim M, Millar AA, Wood CC, Larkin PJ (2012) Resistance to wheat streak mosaic virus generated by expression of an artificial polycistronic microRNA in wheat. Plant Biotechnol J 10:150–163
Gasparis S, Orczyk W, Nadolska-Orczyk A (2013) Sina and Sinb genes in triticale do not determine grain hardness contrary to their orthologs Pina and Pinb in wheat. BMC Plant Biol 13:190
Gasparis S, Kala M, Przyborowski M, Orczyk W, Nadolska-Orczyk A (2017) Artificial microRNA-based specific gene silencing of grain hardness genes in polyploid cereals appeared to be not stable over transgenic plant generations. Front Plant Sci 7:2017
German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, De Paoli E, Lu C, Schroth G, Meyers BC, Green PJ (2008) Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26:941–946
Goto K, Kanazawa A, Kusaba M, Masuta C (2003) A simple and rapid method to detect plant siRNAs using nonradioactive probes. Plant Mol Biol Report 21:51–58
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109–D111
Griffiths-Jones S (2010) miRBase: microRNA sequences and annotation. Curr Protoc Bioinform 29:12.9-1–12.9-10
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34:D140–D144
Inal B, Turktas M, Eren H, Ilhan E, Okay S, Atak M, Erayman M, Unver T (2014) Genome-wide fungal stress responsive miRNA expression in wheat. Planta 240:1287–1298
Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23:4185–4207
Jeong DH, Schmidt SA, Rymarquis LA, Park S, Ganssmann M, German MA, Accerbi M, Zhai JX, Fahlgren N, Fox SE, Garvin DF, Mockler TC, Carrington JC, Meyers BC, Green PJ (2013) Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol 14:R145. http://genomebiology.com/2013/14/12/R145. Accessed 5 Oct 2016
Kantar M, Akpinar BA, Valarik M, Lucas SJ, Dolezel J, Hernandez P, Budak H, International Wheat Genome Sequencing C (2012) Subgenomic analysis of microRNAs in polyploid wheat. Funct Integr Genom 12:465–479
Khraiwesh B, Zhu JK, Zhu JH (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta Gene Regul Mech 1819:137–148
Koyama T, Furutani M, Tasaka M, Ohme-Takagi M (2007) TCP transcription factors control the morphology of shoot lateral organs via negative regulation of the expression of boundary-specific genes in Arabidopsis. Plant Cell 19:473–484
Koyama T, Mitsuda N, Seki M, Shinozaki K, Ohme-Takagi M (2010) TCP transcription factors regulate the activities of ASYMMETRIC LEAVES1 and miR164, as well as the auxin response, during differentiation of leaves in Arabidopsis. Plant Cell 22:3574–3588
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Lanet E, Delannoy E, Sormani R, Floris M, Brodersen P, Crete P, Voinnet O, Robaglia C (2009) Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 21:1762–1768
Laufs P, Peaucelle A, Morin H, Traas J (2004) MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 131:4311–4322
Leclercq M, Diallo AB, Blanchette M (2013) Computational prediction of the localization of microRNAs within their pre-miRNA. Nucleic Acids Res 41:7200–7211
Luan M, Xu M, Lu Y, Zhang L, Fan Y, Wang L (2015) Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 555:178–185
Mallory AC, Dugas DV, Bartel DP, Bartel B (2004) MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr Biol 14:1035–1046
Mallory AC, Bartel DP, Bartel B (2005) MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17:1360–1375
Meng F, Liu H, Wang K, Liu L, Wang S, Zhao Y, Yin J, Li Y (2013) Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol 13:140
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Nag A, King S, Jack T (2009) miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc Natl Acad Sci USA 106:22534–22539
Ori N, Cohen AR, Etzioni A, Brand A, Yanai O, Shleizer S, Menda N, Amsellem Z, Efroni I, Pekker I, Alvarez JP, Blum E, Zamir D, Eshed Y (2007) Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat Genet 39:787–791
Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Remita MA, Lord E, Agharbaoui Z, Leclercq M, Badawi MA, Sarhan F, Diallo AB (2016) A novel comprehensive wheat miRNA database, including related bioinformatics software. Curr Plant Biol 7–8:31–33
Saini HS, Westgate ME (1999) Reproductive development in grain crops during drought. Adv Agron 68(1999):59–96
Saldanha AJ (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics 20:3246–3248
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Sun F, Guo G, Du J, Guo W, Peng H, Ni Z, Sun Q, Yao Y (2014) Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol 14:142
Sunkar R, Jagadeeswaran G (2008) In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol 8:37
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17:196–203
Unver T, Budak H (2009) Conserved microRNAs and their targets in model grass species Brachypodium distachyon. Planta 230:659–669
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Vaucheret H, Vazquez F, Crete P, Bartel DP (2004) The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev 18:1187–1197
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wei B, Cai T, Zhang R, Li A, Huo N, Li S, Gu YQ, Vogel J, Jia J, Qi Y, Mao L (2009) Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat (Triticum aestivum L.) and Brachypodium distachyon (L.) Beauv. Funct Integr Genom 9:499–511
Wei LQ, Yan LF, Wang T (2011) Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol 12:R53
Williams L, Grigg SP, Xie M, Christensen S, Fletcher JC (2005) Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 132:3657–3668
Willmann MR, Poethig RS (2007) Conservation and evolution of miRNA regulatory programs in plant development. Curr Opin Plant Biol 10:503–511
Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y (2010) DNA methylation mediated by a microRNA pathway. Mol Cell 38:465–475
Xin MM, Wang Y, Yao YY, Xie CJ, Peng HR, Ni ZF, Sun QX (2010) Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol 10:123. http://www.biomedcentral.com/1471-2229/10/123. Accessed 5 Oct 2016
Yan Y, Wang H, Hamera S, Chen X, Fang R (2014) miR444a has multiple functions in the rice nitrate-signaling pathway. Plant J 78:44–55
Yang C, Li D, Mao D, Liu X, Ji C, Li X, Zhao X, Cheng Z, Chen C, Zhu L (2013) Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ 36:2207–2218
Yao YY, Guo GG, Ni ZF, Sunkar R, Du JK, Zhu JK, Sun QX (2007) Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol 8:R96. doi:10.1186/gb-2007-8-6-r96
Yu B, Wang H (2010) Translational inhibition by microRNAs in plants. Prog Mol Subcell Biol 50:41–57
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA (2005) Identification and characterization of new plant microRNAs using EST analysis. Cell Res 15:336–360
Zhang BH, Pan XP, Anderson TA (2006a) Identification of 188 conserved maize microRNAs and their targets. FEBS Lett 580:3753–3762
Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA (2006b) Evidence that miRNAs are different from other RNAs. Cell Mol Life Sci 63:246–254
Zhang L, Chia JM, Kumari S, Stein JC, Liu Z, Narechania A, Maher CA, Guill K, McMullen MD, Ware D (2009) A genome-wide characterization of microRNA genes in maize. PLoS Genet 5:e1000716
Zhou ZS, Huang SQ, Yang ZM (2008) Bioinformatic identification and expression analysis of new microRNAs from Medicago truncatula. Biochem Biophys Res Commun 374:538–542
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61:4157–4168
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415
Acknowledgements
This research was supported by the Polish National Science Centre, Grant UMO-2011/03/B/NZ9/01383.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
The research was fully financed by the Polish National Science Centre, Grant UMO-2011/03/B/NZ9/01383.
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by E. Schleiff.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gasparis, S., Yanushevska, Y. & Nadolska-Orczyk, A. Bioinformatic identification and expression analysis of new microRNAs from wheat (Triticum aestivum L.). Acta Physiol Plant 39, 236 (2017). https://doi.org/10.1007/s11738-017-2530-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-017-2530-6