
Abstract
Background
Adavosertib (AZD1775) is a first-in-class, selective, small-molecule inhibitor of Wee1.
Objective
The safety, tolerability, pharmacokinetics, and efficacy of adavosertib monotherapy were evaluated in patients with various solid-tumor types and molecular profiles.
Patients and Methods
Eligible patients had the following: confirmed diagnosis of ovarian cancer (OC), triple-negative breast cancer (TNBC), or small-cell lung cancer (SCLC); previous treatment for metastatic/recurrent disease; and measurable disease. Patients were grouped into six matched cohorts based on tumor type and presence/absence of biomarkers and received oral adavosertib 175 mg twice a day on days 1–3 and 8–10 of a 21-day treatment cycle.
Results
Eighty patients received treatment in the expansion phase; median total treatment duration was 2.4 months. The most common treatment-related adverse events (AEs) were diarrhea (56.3%), nausea (42.5%), fatigue (36.3%), vomiting (18.8%), and decreased appetite (12.5%). Treatment-related grade ≥ 3 AEs and serious AEs were reported in 32.5% and 10.0% of patients, respectively. AEs led to dose interruptions in 22.5%, reductions in 11.3%, and discontinuations in 16.3% of patients. One patient died following serious AEs of deep vein thrombosis (treatment related) and respiratory failure (not treatment related). Objective response rate, disease control rate, and progression-free survival were as follows: 6.3%, 68.8%, 4.5 months (OC BRCA wild type); 3.3%, 76.7%, 3.9 months (OC BRCA mutation); 0%, 69.2%, 3.1 months (TNBC biomarker [CCNE1/MYC/MYCL1/MYCN] non-amplified [NA]); 0%, 50%, 2 months (TNBC biomarker amplified); 8.3%, 33.3%, 1.3 months (SCLC biomarker NA); and 0%, 33.3%, 1.2 months (SCLC biomarker amplified).
Conclusion
Adavosertib monotherapy was tolerated and demonstrated some antitumor activity in patients with advanced solid tumors.
Trial Registration
ClinicalTrials.gov identifier NCT02482311; registered June 2015.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This phase Ib study assessed the safety, tolerability, and antitumor activity of adavosertib monotherapy in patients with advanced solid tumors. |
Adavosertib demonstrated some antitumor activity in heavily pretreated patients with advanced solid tumors. |
The safety and tolerability profile of adavosertib monotherapy was considered manageable. |
1 Introduction
Wee1, a protein tyrosine kinase that phosphorylates and inhibits cyclin-dependent kinase (CDK) 1 and 2, is involved in the regulation of the G2 and S phases of the cell cycle [1,2,3] and is central in maintaining genomic integrity through epigenetic modification of histones and regulation of histone synthesis in late S phase and anaphase onset [4]. Most human cancer cells have mutations in the TP53 gene that result in failure to arrest at the G1/S checkpoint and are dependent on the S and G2 checkpoints for DNA damage repair prior to mitosis [3, 5, 6]. Thus, inhibition of Wee1 is an attractive target for anticancer therapy as a method of stimulating CDK1 and CDK2 activation, uncontrolled DNA replication and replication stress, release of G2 cell-cycle arrest, and premature mitosis of cancer cells [3]. Unrepaired double-strand breaks and under-replicated DNA may in turn lead to mitotic catastrophe and cell death [7]. Abrogation of the G2/M cell-cycle checkpoint also exploits the G1 deficiency in TP53-mutated tumor cells by enhancing the apoptotic response to DNA damage induced by chemotherapy or other anticancer agents [6].
Adavosertib (AZD1775) is an orally active, first-in-class, small-molecule reversible inhibitor of Wee1 kinase in development for the treatment of advanced solid tumors [8]. Early preclinical studies investigated the ability of adavosertib treatment to synergize with chemotherapy [8,9,10]. The antitumor activity of adavosertib when administered in combination with DNA-damaging agents, including gemcitabine, carboplatin, and cisplatin, was greater in p53-deficient tumor cells (vs those with functional p53), supporting the hypothesis that tumors with a defective G1 checkpoint are dependent on the G2 checkpoint to escape mitotic lethality [3, 8, 10]. Promising clinical antitumor activity has subsequently been shown with adavosertib in combination with chemotherapy in phase I studies of patients with advanced solid tumors [11, 12], a phase II trial in locally advanced pancreatic cancer [13], and in early phase II studies in TP53-mutated refractory/resistant ovarian cancer [14] and platinum-refractory/resistant epithelial ovarian cancer [15].
Observations from other preclinical studies suggested that adavosertib may also elicit a p53-independent defective DNA damage response [16, 17]. Indeed, adavosertib monotherapy induces high levels of replication stress, endogenous DNA damage, and defects in DNA damage responses, resulting in mitotic catastrophe and cell death [2, 16, 17]. This was confirmed in a phase I study that, in addition to providing the first direct evidence of a reduction in phosphorylated Tyr15-Cdk (a target of Wee1 kinase) levels in paired tumor biopsies, showed increased levels of phosphorylated histone gamma H2AX (which is indicative of a DNA damage response) [18]. Adavosertib showed single-agent activity in patient-derived xenograft mouse models and/or cell lines for ovarian cancer [19], triple-negative breast cancer (TNBC) [19, 20], and small-cell lung cancer (SCLC) [21]. Adavosertib may be particularly effective in tumors with selected genetic biomarkers, including those that are indicative of an impaired DNA damage response (BRCA1/2 mutation), replication stress caused by oncogenic drivers (MYC/MYCL1/MYCN amplification), or cell-cycle dysregulation (cyclin E1 [CCNE1] amplification) [2, 22]. Preliminary antitumor activity has been demonstrated with adavosertib monotherapy in phase I studies involving patients with advanced solid tumors [23, 24] and BRCA-proficient and BRCA-deficient ovarian and endometrial cancer [18, 25].
The current phase Ib study (ClinicalTrials.gov identifier NCT02482311) assessed the safety and tolerability of adavosertib monotherapy in patients with advanced solid tumors, as well as the antitumor activity of adavosertib monotherapy in previously treated patients with ovarian cancer, TNBC, or SCLC. Cohorts were based on tumor type and molecular profile (BRCA status, CCNE1 amplification, or MYC/MYCL1/MYCN amplification). Key secondary objectives were to characterize the pharmacokinetic (PK) profile of adavosertib and to identify genetic biomarkers that correlate with clinical outcomes.
2 Methods
2.1 Study Design and Treatment Schedule
This open-label, multicenter, phase Ib study was designed to evaluate the safety, tolerability, PKs, and antitumor activity of oral adavosertib monotherapy in patients with advanced solid tumors. The study was divided into two parts (Fig. 1). Part A was a safety lead-in, in a cohort of patients with advanced solid tumors; in part B, the expansion cohorts, patients were grouped based on tumor type (ovarian cancer, TNBC, or SCLC) and molecular profile (BRCA status, CCNE1 amplification, or MYC/MYCL1/MYCN amplification) to evaluate antitumor activity. Patients in the safety lead-in cohort received oral adavosertib (200 mg twice a day [bid]) on days 1–3 and 8–10 of a 21-day cycle. Following observation of diarrhea, vomiting, and anemia during the safety lead-in, the dosage was lowered from 200 mg bid to 175 mg bid and mandatory anti-emetic prophylaxis (consisting of a serotonin 5-HT3 antagonist, ondansetron 8 mg bid, or granisetron 1 mg bid prior to each dose of adavosertib plus dexamethasone 4 mg with each adavosertib dose as a minimum on the first day of dosing of every 3-day dosing period) was instituted. Patients in the expansion cohorts received oral adavosertib (175 mg bid) on days 1–3 and 8–10 of a 21-day treatment cycle.

Study design. All tumor samples were analyzed for a range of cancer-related genes such that the clinical response could be correlated with the genetic aberration. BRCA wild type was defined as no evidence of deleterious or suspected deleterious mutation in the BRCA1 or BRCA2 genes. BRCA1 and/or BRCA2 variants classified as a ‘variant of uncertain clinical significance’ or ‘variant of unknown significance,’ as well as ‘variant, favors polymorphism’ or ‘benign polymorphism,’ were considered to be BRCA wild type. Biomarker-positive TNBC and SCLC cohorts were defined as having amplifications in CCNE1, MYC, MYCL1, or MYCN. Biomarker-negative TNBC and SCLC cohorts were defined as the absence of the qualifying amplifications for TNBC or SCLC specified above. bid twice a day, BRCAm breast cancer gene 1/2 mutation, BRCAwt breast cancer gene 1/2 wild type, CCNE1 cyclin E1, n number of patients, PARPi poly(ADP-ribose) polymerase inhibitor, SCLC small-cell lung cancer, TNBC triple-negative breast cancer
These patients were assigned to one of six subgroups based on tumor type and molecular profile. These subgroups included patients with the following: BRCA wild-type (BRCAwt) ovarian cancer; BRCA-associated ovarian cancer (BRCAm) with documented poly(ADP-ribose) polymerase inhibitor (PARPi) failure; TNBC with amplification of CCNE1 or MYC/MYCL1/MYCN; TNBC with non-amplified status for these genes; SCLC with amplification of CCNE1 or MYC/MYCL1/MYCN; and SCLC with non-amplified status for these genes. Amplification status and BRCA status were assessed by tumor testing of archival tissue, performed by a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory in Foundation Medicine (FMI). CCNE1 and MYC/MYCL1/MYCN amplification status was determined in accordance with the FMI amplification definition. Based on a model-determined estimated tumor cell ploidy, the threshold for calling an amplification was ploidy plus four. If a known amplification was found to be non-focal, the amplification was reported as ambiguous rather than confirmed. In both patient cohorts, treatment continued until disease progression or unacceptable toxicity; disease restaging was performed every 6 weeks (± 7 days) in the first year and every 12 weeks thereafter (expansion cohorts only). The study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on bioethics [26]. The institutional review boards or independent ethics committees of all investigational sites approved the protocol, and all patients provided written informed consent.
2.2 Patients
Patients enrolled in the study were required to be at least 18 years old and have the following: measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (part B only); an Eastern Cooperative Oncology Group performance status of 0 or 1; and prior receipt of chemotherapy for recurrent or metastatic disease. To be eligible for inclusion in part A, patients had to have a histologically or cytologically documented locally advanced or metastatic solid tumor, excluding lymphoma, for which standard therapy did not exist or had proven ineffective or intolerable. To be eligible for inclusion in part B, patients were required to have had a histologically confirmed diagnosis of epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer refractory to standard therapies or for which no standard therapy existed (BRCAm [status determined from archival tissue or reported by investigator site; germline BRCA status was not known in all cases] with documented disease progression during or after treatment with a PARPi, or biomarker negative [BRCAwt] and completed more than three prior lines of chemotherapy), TNBC, or SCLC. Patients with TNBC, defined as minimal or no expression of estrogen, progesterone, and human epidermal growth factor receptor 2 (HER2) receptors with biomarker-positive or biomarker-negative tumors (CCNE1 and/or MYC/MYCL1/MYCN amplification), could have received one or more prior lines of chemotherapy. Patients with SCLC and biomarker-negative or biomarker-positive tumors (CCNE1 and/or MYC/MYCL1/MYCN amplification) could have received no more than one prior line of chemotherapy with documented disease relapse 90 or more days following that treatment. Where data were available, the baseline tumor grade and histology type by subgroup are indicated in Table 1.
Key exclusion criteria included the use of any anticancer treatment drug within the last 21 days or five half-lives (whichever was shorter) prior to the first administration of study drug and use of other anticancer therapy (except palliative local radiotherapy); biological therapy or other novel agent during study treatment; concomitant use of medications or other products with known inducer or inhibitory effects on cytochrome P450 (CYP) 3A4; and the inability to swallow oral medications.
2.3 Primary Outcome Measures
2.3.1 Safety and Tolerability
In part A, dose-limiting toxicities (DLTs) were recorded and the maximum tolerated dose (MTD) was determined. DLTs were defined as any of the following toxicities not attributable to disease or disease-related processes under investigation: hematological toxicity grade ≥ 4 for > 7 days, including infection with neutropenia; grade 3 thrombocytopenia associated with grade ≥ 2 bleeding; non-hematological toxicity grade ≥ 3; grade ≥ 3 total bilirubin (> 2.5–5 × upper limit of normal [ULN]), alanine aminotransferase (> 5–10 × ULN), aspartate aminotransferase (> 5–10 × ULN), or alkaline phosphatase (> 5–10 × ULN) lasting > 48 h; changes in liver function tests; any other toxicity that was clinically significant and/or unacceptable, which did not respond to supportive care and resulted in a disruption of dosing schedule of more than 7 days, or was judged to be a DLT by the investigator (in collaboration with the medical monitor). Primary safety and tolerability outcome measures were treatment-emergent adverse events (AEs), graded by Common Terminology Criteria for Adverse Events (CTCAE; version 4.03) and assessed by physical examination, vital signs, and laboratory parameters.
Adavosertib 175 mg bid was selected for dose expansion as previously described [27]. In summary, 12 patients were recruited into the safety lead-in cohort. Seven patients were treated at the initial dosage of 200 mg bid, and three patients experienced DLTs, leading to a dosage reduction to 175 mg bid. The five additional patients in the safety lead-in cohort were treated with the initial dosage of adavosertib 175 mg bid, with the addition of antiemetic prophylaxis, and one patient experienced a DLT. The sixth consecutive patient to receive the initial dosage of 175 mg bid did not experience a DLT, and was included in part B (ovarian cancer, BRCAm, PARPi failure expansion cohort).
2.3.2 Efficacy
Primary efficacy outcome measures included the following: objective response rate (ORR), defined as confirmed complete response (CR) plus confirmed partial response (PR); disease control rate (DCR), defined as the proportion of patients with a confirmed best objective response (BOR) of CR or PR, or BOR of stable disease; duration of response (DOR); and progression-free survival (PFS) based on RECIST version 1.1.
2.4 Secondary Outcome Measures
2.4.1 Pharmacokinetic Assessments
Blood samples for adavosertib PK assessments were collected during the part A safety lead-in phase on cycle 1 day 1, and day 3 or 10 at predefined time points. During part B, optional blood samples were taken from consenting patients on days 3 or 10 of cycle 1, cycle 2, or cycle 4 and, beyond cycle 4, every two cycles at predefined time points. Key PK parameters evaluated were area under the plasma concentration–time curve from time zero to 12 h (AUC12); maximum plasma drug concentration (Cmax); plasma drug concentration at 8 h (C8); lowest plasma drug concentration (Ctrough); time to reach maximum plasma concentration (tmax); and apparent terminal elimination (t½λz).
2.4.2 Biomarker Assessments
Patients provided additional informed consent for the optional collection of genetic material from archival tumor tissue. Archival tumor tissue was provided in formalin-fixed paraffin-embedded (FFPE) blocks; if FFPE blocks were unavailable, archival tumor tissue sections were provided on tissue slides; all tissue was shipped at ambient temperature to a central laboratory for processing. Profiling of somatic genetic alterations in the archival tumors of patients responding to adavosertib treatment was performed using the FoundationOne® assay (F1) and analyzed using FMI’s F1 classification rules; targeted genomic profiling was presented using an in-house bioinformatics platform (Oncoprofile tool) and correlated with clinical outcomes [28].
2.5 Statistical Analyses
Up to 172 patients were planned to be enrolled and treated in this study, 12 in the part A safety lead-in cohort and a maximum of up to 160 in part B expansion cohorts (ovarian [n = 60], TNBC [n = 50]), and SCLC [n = 50]). Descriptive statistics were used to summarize the safety, PK, and antitumor activity data. Statistical analyses were performed using SAS® (SAS Institute, Cary, NC, USA) by SC Innovations under the direction of the Biometrics Group, AstraZeneca. The full analysis set included all patients (in the part A safety lead-in and part B expansion cohort) treated with at least one dose of adavosertib; this population was used for the primary analyses of the safety and efficacy endpoints. The dose-limiting analysis set (part A only) included all patients in the safety lead-in cohort who received at least 75% of the adavosertib dose (at least nine of 12 doses) and completed the minimum safety requirements during the 21 days of treatment or who experienced a DLT during the first 21 days of treatment regardless of the number of doses received. The PK analysis set included all patients who received at least one dose of adavosertib and provided at least one PK sample. The biomarker analysis set included all patients who consented and provided a valid archived tumor sample.
3 Results
3.1 Part A: Safety Lead-In Cohort
3.1.1 Patients
A total of 12 patients were enrolled, seven of whom received adavosertib 200 mg bid orally and five of whom received adavosertib 175 mg bid orally. Baseline characteristics of the patients enrolled in part A are provided in Table 1. All 12 patients discontinued treatment, the majority (66.7%) due to disease progression (Supplementary Figure S1, see the electronic supplementary material).
3.1.2 Safety
All seven patients enrolled in the 200-mg cohort experienced a treatment-emergent AE, the most common of which were nausea (100%), diarrhea (85.7%), vomiting (85.7%), and fatigue (71.4%). Grade ≥ 3 treatment-emergent AEs occurred in one patient (14.3%) and included events of hypovolemia and anemia that were considered treatment related. Five patients were enrolled in the 175-mg safety lead-in cohort, of whom 80% experienced a treatment-emergent AE, including fatigue (60%), vomiting (60%), diarrhea (40%), and nausea (40%). Except for fatigue, all were considered treatment related. In the 175-mg cohort, grade ≥ 3 treatment-emergent AEs occurred in one patient (20%) and included a serious AE (SAE) of non-treatment-related abdominal pain and mental status changes attributed to underlying disease (SCLC).
Four patients experienced a total of eight DLTs, all of which were grade 3. In the 200-mg treatment group, three patients experienced five DLTs: one patient had hypovolemia, the second had diarrhea, and the third had diarrhea, nausea, and vomiting. In the 175-mg treatment group, one patient experienced three DLTs: diarrhea, nausea, and vomiting. The MTD of adavosertib was established as 175 mg bid on days 1–3 and 8–10 of a 21-day treatment cycle. This dosage was used to treat patients in the part B expansion cohort.
3.2 Part B: Expansion Cohorts
3.2.1 Patients
A total of 82 patients were enrolled, and 80 (97.6%) received adavosertib treatment. Patients had a median (range) age of 60 (35–83) years, and 73 (91.3%) were women. Baseline characteristics for patients grouped by tumor type and molecular profile are summarized in Table 1. Of the 80 patients who received adavosertib, two (2.5%) remained on treatment at the time of data cut-off. Of the 78 (97.5%) patients who discontinued treatment, 56 (70%) discontinued because of disease progression (Supplementary Figure S1, see the electronic supplementary material). Three patients (3.8%) discontinued treatment for other reasons as reported by investigators; two (2.5%) because of clinical progression; and one (1.3%) because of an increased worsening of AEs and an increase in tumoral markers.
3.2.2 Safety
Treatment-related AEs were experienced by 67 patients (83.8%); those occurring in 10% or more of the patients included diarrhea (45 patients [56.3%]), nausea (34 [42.5%]), fatigue (29 [36.3%]), vomiting (15 [18.8%]), and decreased appetite (10 [12.5%]). Treatment-related AEs occurring in the blood and lymphatic system included anemia, neutropenia, and thrombocytopenia in eight (10.0%), five (6.3%), and five patients (6.3%), respectively. Treatment-related AEs of grade 3 or above were reported in 26 patients (32.5%) in part B. Table 2 shows a summary of the most common treatment-related AEs by event for the full analysis set, part A safety lead-in cohort, part B expansion cohorts, and grade ≥ 3 treatment-related AEs for the full analysis set.
SAEs were reported in 18 patients (22.5%), eight (10.0%) of whom experienced a total of 12 SAEs that were considered causally related to study treatment by the investigator: vomiting (n = 2), dehydration (n = 2), anemia, nausea, sepsis, urinary tract infection, platelet count decreased, syncope, hypoxia, and deep vein thrombosis (DVT). The single patient who died from an SAE of DVT (assessed to be treatment related by the investigator) also experienced an SAE of non-treatment-related respiratory failure. There were no other deaths due to AEs.
AEs (treatment related and treatment emergent) led to dose interruptions in 18 patients (22.5%), dose reductions in nine (11.3%), and discontinuations in 13 (16.3%); of these, interruptions were attributed to gastrointestinal disorders (most commonly nausea and vomiting) in ten patients (12.5%), and reductions were attributed to abdominal pain, diarrhea, nausea, and vomiting in five patients (6.3%). No patients discontinued treatment because of diarrhea, nausea, or vomiting; however, four patients (5.0%) discontinued because of other gastrointestinal disorders (e.g., abdominal pain, ileal perforation, or intestinal obstruction). Treatment was interrupted in one patient (1.3%) who experienced anemia. There were no dose reductions associated with AEs in the blood and lymphatic system; however, one patient (1.3%) developed thrombocytopenia, which led to discontinuation of treatment. Overall, the median (range) treatment duration for the part B expansion cohort was 2.4 (0–17.4) months.
3.2.3 Efficacy
Responses to adavosertib treatment are summarized in Table 3. Adavosertib demonstrated some antitumor activity in patients in the ovarian cancer cohorts (both the BRCAwt and the BRCAm with PARPi failure cohorts), and the biomarker-negative SCLC cohort. The ORR for all patients in the part B expansion cohort was 3.8% (three patients; 95% confidence interval [CI] 0.8–10.6). The DCR was 63.8% (51 patients; 95% CI 52.2–74.2). A confirmed PR was observed in three patients: one in the ovarian cancer, BRCAwt cohort (DOR 13.9 months, censored at the time of data cut-off); one in the ovarian cancer, BRCAm, PARPi failure cohort (DOR 4.7 months); and one in the SCLC, biomarker-negative cohort (DOR 6.9 months). The number of confirmed PRs meant that calculation of DOR would not be robust. A total of 56 treated patients (60.9%) in the study overall had a BOR of stable disease for ≥ 5 weeks, including six patients (6.5%) with an unconfirmed PR. At their second scheduled post-baseline tumor assessment (12 weeks), a total of 30 treated patients (37.5%) in the part B expansion cohort had a BOR of stable disease, the majority of whom were in the ovarian cancer, BRCAm, PARPi failure or the ovarian cancer, BRCAwt cohorts (13 and eight patients, respectively). Median PFS was 3.0 months (95% CI 2.6–4.1) across all tumor types. Median PFS was 4.5 months for patients in the ovarian cancer, BRCAwt cohort (95% CI 1.5–7.2); 3.9 months in the ovarian cancer, BRCAm, PARPi failure cohort (95% CI 2.6–5.8); 3.1 months in the TNBC, biomarker-negative cohort (95% CI 1.4–5.3); 2.0 months in the TNBC, biomarker-positive cohort (95% CI 1.3–4.1); 1.3 months in the SCLC, biomarker-negative cohort (95% CI 1.1–5.1); and 1.2 months in the SCLC, biomarker-positive cohort (95% CI 1.2–3.8). PFS across all tumor types and percentage change from baseline of target lesions at ≥ 5 weeks by cohort are shown in Supplementary Figures S2 and S3 (see the electronic supplementary material). Greater changes in lesion size were observed in the ovarian and SCLC cohorts than in the TNBC cohorts (Supplementary Figure S3).
3.2.4 Pharmacokinetics
Key PK parameters are summarized in Table 4. Adavosertib was steadily absorbed and slowly eliminated following once- or twice-daily administration of single or multiple oral doses (175 mg or 200 mg). Median tmax was 2–3 h, and mean t½λz was 5–7 h. After oral administration of 175 mg or 200 mg of adavosertib, systemic exposure increased in a near dose-proportional manner. Adavosertib accumulated in the plasma (based on AUC12) with geometric mean accumulation ratios of approximately 2.1. Clinical PKs of 175 mg bid (day 3 exposure) suggested that concentrations were above the target pCDK1 half-maximal inhibitory concentration (IC50) of 240 nmol/L and within the cell kill zone (500–1000 nmol/L) derived from non-clinical data (Supplementary Figure S4, see the electronic supplementary material).
3.2.5 Biomarker Analysis
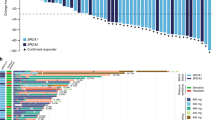
Results from patients with available next-generation sequencing (NGS) data are summarized in Fig. 2. A total of 85 patients who received treatment had successful NGS performed on archival tumor tissue. The TP53 mutation was the most common genetic aberration and was observed in 82% of all patients regardless of tumor type. In patients with ovarian cancer, mutations in BRCA1 were more common than mutations in BRCA2 (observed in 41% and 14% of patients, respectively). CCNE1 amplification was detected in the archival tumors of three patients (one patient each in the ovarian cancer, BRCAwt; ovarian cancer, BRCAm, PARPi failure; and TNBC, CCNE1/MYC/MYCL1/MYCN amplified cohorts), who all experienced a reduction in target lesion size. Matched pre- and post-treatment samples to assess alteration status in matched samples were unavailable. Of the 27 patients with available NGS data in the BRCAm, PARPi failure cohort, 21 (78%) had an alteration detected in BRCA1 or BRCA2, while six (22%) did not have a deleterious alteration detected in the archival tumor tissue sample.

FoundationOne® genomic profiles of archival tumor tissue and clinical outcomes across patient cohorts: A all patients who received adavosertib with NGS and response data (n = 85); B–D part B expansion cohorts; B ovarian cancer expansion cohorts (n = 42 [BRCAwt, n = 15/16; BRCAm, PARPi failure, n = 27/30]); C TNBC expansion cohorts (n = 18/19); D SCLC expansion cohorts (n = 14/15). The 60 most frequently detected altered cancer-related genes are displayed in A–C; the expansion cohorts in B–D are a subset of A; only 40 altered cancer-related genes were detected in the SCLC expansion cohorts shown in (D). Best change (%) best change in target lesion size is the maximum reduction from baseline or the minimum increase from baseline in the absence of a reduction, bid twice a day, BOR best overall response, BRCAm breast cancer gene 1/2 mutation, BRCAwt breast cancer gene 1/2 wild type, CCNE1 cyclin E1, FS frameshift, n number of patients, NE not evaluable, NGS next-generation sequencing, PARPi poly(ADP-ribose) polymerase inhibitor, PD progressive disease, PFS progression-free survival, PR partial response, SCLC small-cell lung cancer, SD stable disease, TNBC triple-negative breast cancer, TRT treatment, Trunc truncation, VUS variation of unknown significance
Too few clinical responses were observed to be able to correlate response with biomarker status based on the available NGS data.
4 Discussion
This open-label, multicenter, phase Ib study evaluated the safety, tolerability, and antitumor activity of adavosertib monotherapy in patients with advanced solid tumors.
Safety data were consistent with the known safety profile of adavosertib monotherapy [18, 23]. The most common treatment-related AEs, including those with a CTCAE grade of 3 or above, were generally related to the gastrointestinal system. Nausea and vomiting are part of the known toxicity profile of adavosertib, and as such, all patients received mandatory antiemetic prophylaxis in accordance with the study protocol. However, the fact that gastrointestinal disorders remained among the most common reasons for dose interruptions or reductions in this study suggests that further efforts may be required to optimize the pre-medication antiemetic regimen. Although hematologic toxicities associated with adavosertib treatment resulted in two treatment interruptions and one discontinuation, no dose reductions were required. In the case of treatment-related DVT and respiratory failure that led to death, the investigator suspected pulmonary embolism as the cause of death, although a confirmatory computed tomography scan was not performed.
An overall DCR of 63.8% was observed across all cohorts, which included patients who had been previously treated for ovarian cancer (including those with BRCA-mutated ovarian cancer who had previously failed to respond to PARPi therapy), TNBC, or SCLC. The antitumor activity observed in the current study builds on the existing evidence base with adavosertib monotherapy. In a previous phase Ib study designed to determine the maximum tolerated dosing schedule for adavosertib monotherapy, preliminary antitumor activity was observed in heavily pretreated patients with locally advanced or metastatic solid tumors, with a safety profile that was acceptable and manageable [23]. Adavosertib monotherapy has also demonstrated clinical activity in women with recurrent uterine serous carcinoma and will be further investigated in this population in the ongoing phase IIb ADAGIO trial [29, 30]. Furthermore, antitumor activity has been demonstrated following daily adavosertib monotherapy in a phase I study in patients with BRCA-proficient and BRCA-deficient ovarian and endometrial cancer; the elevated pY15-Cdk levels observed support the hypothesized cell cycle mechanism of action and may provide an important correlate of response [18, 25]. In accordance with these findings, patients in both ovarian cancer cohorts in the present study had the longest median PFS and highest DCR of all patients in part B, and two of the three confirmed PRs were also observed in patients with ovarian cancer.
The present study also offers further insights into the PK properties of adavosertib. Preclinical studies have indicated that higher adavosertib exposure, longer duration, and greater number of days of dosing lead to increased antitumor activity [31]. At the time of study protocol development, previous investigation of adavosertib monotherapy had been limited to either single dosing before combination treatment or using 225 mg bid for five doses over 2.5 days in a small cohort of patients with refractory tumors [18]. The dosing schedule chosen for this study extended the dosing of adavosertib to six doses over 3 days. The original dosing was 200 mg bid over 3 days, which approximately equated to the 2.5 days schedule of the previously tested 225-mg bid dosing and was considered more practical. Although the adavosertib concentrations measured in this study were above the target range predicted based on preclinical data, it is hypothesized that this dosing level remained suboptimal and could be the reason for the limited clinical activity observed. A subsequent study to maximize the exposure and increase the clinical benefit of adavosertib investigated the maximum tolerated dosing schedules for adavosertib monotherapy (ClinicalTrials.gov identifier NCT02610075). Adavosertib monotherapy showed preliminary efficacy in heavily pretreated patients with locally advanced or metastatic solid tumors, with a safety profile that was acceptable and manageable. The bid MTD was determined to be 125 mg (5/9 schedule [5 days on treatment followed by 9 days off]), and the once daily (qd) MTD was 300 mg (5/2 schedule or 5/9 schedule) for 2/3 weeks. The recommended phase II dose was 300 mg (qd; 5/2 schedule) [23], which was utilized in both the NCT03668340 and the subsequent ADAGIO phase II studies in patients with uterine serous carcinoma [29, 30].
Inhibiting Wee1 kinase to compromise the G2/M checkpoint may potentiate the effectiveness of chemotherapy [32]; indeed, the safety and efficacy of adavosertib when administered in combination with chemotherapy is also currently under clinical investigation. Phase I studies in patients with advanced solid tumors have demonstrated the safety and tolerability of adavosertib plus chemotherapy [11, 12]. Phase II data have shown adavosertib to have promising antitumor activity in TP53-mutated refractory/resistant ovarian cancer when combined with carboplatin [14]. In women with recurrent, platinum-refractory/-resistant epithelial ovarian cancer, adavosertib in combination with gemcitabine was compared with gemcitabine monotherapy in a separate phase II trial; combination therapy improved the response rate, PFS, and overall survival (OS) with manageable toxicity. Correlative analyses to identify potential predictive biomarkers of response are ongoing [15]. Promising early data have also been observed in a phase II trial in patients with locally advanced pancreatic cancer, where adavosertib in combination with gemcitabine and radiation was well tolerated and resulted in substantially higher OS than previous trials combining gemcitabine and radiation [13]. Several trials are ongoing to investigate these preliminary findings further, including a phase II trial of adavosertib in combination with four different chemotherapy agents in patients with ovarian cancer [33] and phase I studies of adavosertib in combination with durvalumab or olaparib in patients with advanced solid tumors [34, 35].
There is some evidence that adavosertib may show greater efficacy against tumors with selected genetic biomarkers [19], including those that are indicative of an impaired DNA damage response (BRCA1/2 mutation), replication stress caused by oncogenic drivers (MYC/MYCL1/MYCN amplification), or cell-cycle dysregulation (CCNE1 amplification) [2, 22]. While all three patients with CCNE1 amplification had reductions in target lesion size, the limited number of clinical responses prevented correlation with biomarker status. Further studies to determine biomarkers of clinical efficacy following treatment with adavosertib are warranted. Retrospective NGS results from archival tumor tissue did not always concur with investigator-reported BRCA status. One of three patients with a CCNE1 amplification was from the BRCAm PARPi failure cohort; however, NGS did not detect any BRCA mutation in the archival tumor tissue sample.
Limitations of this study included the small number of patients in each cohort and, as such, there is a need for further studies to evaluate the efficacy of adavosertib monotherapy in ovarian cancer. The biomarker analysis strategy focused on identification of oncogene alterations (e.g., amplification), and the potential effect of protein overexpression (± oncogene alterations) on tumor sensitivity to Wee1 inhibition was not investigated [36].
5 Conclusions
Adavosertib demonstrated some antitumor activity in heavily pretreated patients with advanced solid tumors but needs to be evaluated in the context of other emerging treatment options. Overall, the safety and tolerability profile of adavosertib monotherapy was considered manageable for patients with advanced solid tumors for which standard therapy does not exist or has proven ineffective or intolerable.
References
Gerard C, Goldbeter A. The balance between cell cycle arrest and cell proliferation: control by the extracellular matrix and by contact inhibition. Interface Focus. 2014;4(3):20130075.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–60.
Do K, Doroshow JH, Kummar S. WEE1 kinase as a target for cancer therapy. Cell Cycle. 2013;12(19):3159–64.
Fu S, Wang Y, Keyomarsi K, Meric-Bernstam F, Meric-Bernstein F. Strategic development of AZD1775, a Wee1 kinase inhibitor, for cancer therapy. Expert Opin Investig Drugs. 2018;27(9):741–51.
Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–7.
Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61(22):8211–7.
Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–37.
Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8(11):2992–3000.
Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9(7):514–22.
Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17(9):2799–806.
Kato H, De Souza P, Kim S-W, Lickliter J, Naito Y, Park K, et al. Safety, pharmacokinetics and clinical activity of adavosertib in combination with chemotherapy in Asian patients with advanced solid tumors: Phase Ib study. Target Oncol. 2020;15(1):75–84.
Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36):4371–80.
Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose escalation trial of the Wee1 INHIBITOR ADAVOSERTIB (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37(29):2643–50.
Leijen S, van Geel R, Sonke G, de Jong D, Rosenberg E, Marchetti S, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36):4354–61.
Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397(10271):281–92.
Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. 2013;12(8):1442–52.
Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11(1):174–82.
Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a WEE1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol. 2015;33(30):3409–15.
O’Connor M, Odedra R, Palakurthi S, Hughes A, Lai Z, Kirschmeier P, et al. Antitumor activity of the WEE1 inhibitor AZD1775 as a monotherapy and in combination with the PARP inhibitor olaparib in patient-derived explant (PDX) models. Eur J Cancer. 2016;69(Suppl 1):S142.
Ha D-H, Min A, Kim S, Jang H, Kim SH, Kim H-J, et al. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci Rep. 2020;10(1):9930.
Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, et al. The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res. 2018;24(20):5153–64.
Naqash AR, Mittra A, Coyne GHOS, Chen L, Das B, Kummar S, et al. Tumor genomic analysis for biomarker identification in a phase I trial of the Wee 1 inhibitor adavosertib (AZD1775) [abstract]. J Clin Oncol. 2020;38(15_suppl):3624.
Falchook G, Sachdev J, Imedio E, Kumar S, Mugundu G, Chmielecki J, et al. A Phase Ib study of WEE1 inhibitor adavosertib in patients with advanced solid tumors [abstract no. CT022]. Cancer Res. 2019;79(13 Suppl):CT022.
Bauer T, Moore K, Rader J, Simpkins F, Mita A, Beck J, et al. Open-label, multicenter, Phase Ib study to assess safety, tolerability and efficacy of adavosertib monotherapy in patients with advanced solid tumors: expansion cohorts [abstract no. CT012]. Cancer Res. 2019;79(13 Suppl):CT012.
Mittra A, Coyne GO, Sullivan, Kummar S, Do K, Bruns A, et al. DNA damage response and therapeutic activity following once-daily administration of the Wee 1 inhibitor AZD1775 (adavosertib) [abstract no. CT099] Cancer Res. 2019;79(13 Suppl):CT099.
AstraZeneca. Bioethics: AstraZeneca global policy. https://www.astrazeneca.com/content/dam/az/PDF/2019/Bioethics%20Policy%20final.pdf. Accessed 16 Mar 2023.
Bauer T, Jones S, Greenlees C, Cook C, Mugundu G, Jewsbury P, et al. Abstract CT013: A phase Ib, open-label, multicenter study to assess the safety, tolerability, pharmacokinetics, and antitumor activity of AZD1775 monotherapy in patients with advanced solid tumors: initial findings. Cancer Res. 2016;76:CT013-CT.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Liu JF, Xiong N, Campos SM, Wright AA, Krasner C, Schumer S, et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J Clin Oncol. 2021;39(14):1531–9.
Liu J, Oza AM, Colombo N, Oaknin A. ADAGIO: a phase IIb international study of the Wee1 inhibitor adavosertib in women with recurrent or persistent uterine serous carcinoma. Int J Gynecol Cancer. 2022;32(1):89–92.
Yates JWT, Cadogan E, Hare JI, Hughes AM, Polanska UM, O'Connor MJ, et al. Analysis of the dose and schedule dependence of tumor kill in nonclinical tumour models after treatment with the WEE1 inhibitor AZD1775 [abstract no. 4302]. Cancer Res. 2018;78(13 Suppl):4302.
Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 2016;37(10):872–81.
Moore K, Chambers S, Hamilton E, Chen L-M, Oza AM, Ghamande S, et al. Adavosertib with chemotherapy (CT) in patients (pts) with platinum-resistant ovarian cancer (PPROC): an open label, four-arm, Phase II study [abstract no. 5513]. J Clin Oncol. 2019;37(Suppl):5513.
Patel MR, Falchook GS, Wang JS-Z, Imedio E, Kumar S, Motlagh P, et al. Open-label, multicenter, Phase I study to assess safety and tolerability of adavosertib plus durvalumab in patients with advanced solid tumors [abstract no. 2562]. J Clin Oncol. 2019;37(Suppl):2562.
Hamilton E, Falchook G, Wang J, Fu S, Oza A, So K, et al. Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: dose escalation [abstract no. CT025]. Cancer Res. 2019;79(13 Suppl):CT025.
Xu H, George E, Kinose Y, Kim H, Shah JB, Peake JD, et al. CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models. Cell Rep Med. 2021;2(9): 100394.
Acknowledgements
We thank the patients who participated in this trial and their families. Medical writing assistance, in the form of the preparation and revision of this article, was supported financially by AstraZeneca and provided by C.L. Attwell Ph.D. of AMICULUM Limited.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was funded by AstraZeneca, which participated in the following: design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflicts of interest
T.M. Bauer has received fees for consulting or advisory roles for Guardant Health, Ignyta (Inst), Loxo, Moderna Therapeutics (Inst), and Pfizer. K. Moore has received fees for advisory boards from AstraZeneca, Aravive, Clovis, Immunogen, Genentech/Roche, GSK/Tesaro, Mersana, Vavotar, Samumed, Mereo, Merck, Pfizer, Janssen, Oncomed, and VBL Therapeutics and is a principal investigator for investigator-initiated clinical trials funded by AstraZeneca and Immunogen. F. Simpkins has participated in advisory boards for AstraZeneca and is a principal investigator for an investigator-initiated clinical trial funded by AstraZeneca. J.T. Beck has received grant funding from AstraZeneca, Bristol-Myers Squibb, Lilly, Novartis, Genentech, AbbVie, Seattle Genetics, Pfizer, and Johnson & Johnson. Q. Chu has received honoraria (unrelated to adavosertib) from and participated in advisory boards for AstraZeneca. A. Oza has participated in advisory boards for AstraZeneca, GlaxoSmithKline, Tesaro, and Clovis and is a principal investigator and has participated in steering committees for clinical trials funded by AstraZeneca. A.V. Tinker has received research funding and honoraria from AstraZeneca and has participated in advisory boards and received honoraria from GlaxoSmithKline. S. Kumar and S. Jenkins are employees and shareholders of AstraZeneca UK Limited. At the time of the study, E. Rodrigo Imedio, G. Mugundu, and J. Chmielecki were employees and shareholders of AstraZeneca UK Limited. E. Rodrigo Imedio and G. Mugundu are employees and shareholders of Takeda. D. Spigel reports grants and/or other from Genentech/Roche, Novartis, Celgene, Bristol-Myers Squibb, AstraZeneca, Pfizer, Boehringer Ingelheim, AbbVie, Foundation Medicine, GlaxoSmithKline, Lilly, Merck, Moderna Therapeutics, Nektar, Takeda, Amgen, TRM Oncology, Precision Oncology, Evelo Therapeutics, Illumina, PharmaMar, University of Texas Southwestern Medical Center Simmons Cancer Center, G1 Therapeutics, Neon Therapeutics, Celldex, Clovis Oncology, Daiichi Sankyo, EMD Serono, Acerta Pharma, Oncogenex, Astellas Pharma, GRAIL, Transgene, Aeglea Biotherapeutics, Tesaro, Ipsen, ARMO BioSciences, Millennium, Genzyme, Intuitive Surgical, Purdue Pharma, Spectrum Pharmaceuticals, and Sysmex. S. Fu, J.S. Rader, A.C. Mita, L. Hart, and S. Jones declare that they have no conflicts of interest.
Compliance and ethical standards
The study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, applicable regulatory requirements, and the AstraZeneca policy on bioethics. The institutional review boards or independent ethics committees of all investigational sites approved the protocol, and all patients provided written, informed consent.
Consent to participate
Written informed consent was obtained for all patients.
Consent for publication
Not applicable.
Availability of data and material
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure. Data for studies directly listed on Vivli can be requested through Vivli at www.vivli.org. Data for studies not listed on Vivli could be requested through Vivli at https://vivli.org/members/enquiries-about-studies-not-listed-on-the-vivli-platform/. An AstraZeneca Vivli member page is also available, outlining further details: https://vivli.org/ourmember/astrazeneca/.
Code availability
Not applicable.
Author contributions
TMB: Resources, data curation, writing—original draft, writing—review and editing. KNM: Resources, data curation, writing—original draft, writing—review and editing. JSR: Resources, data curation, writing—original draft, writing—review and editing. FS: Resources, data curation, writing—original draft, writing—review and editing. ACM: Resources, data curation, writing—original draft, writing—review and editing. JTB: Resources, data curation, writing—original draft, writing—review and editing. LH: Resources, data curation, writing—original draft, writing—review and editing. QC: Resources, data curation, writing—original draft, writing—review and editing. AO: Resources, data curation, writing—original draft, writing—review and editing. AVT: Resources, data curation, writing—original draft, writing—review and editing. ERI: Conceptualization, supervision, methodology, project administration, writing—original draft, writing—review and editing. SK: Conceptualization, supervision, methodology, project administration, writing—original draft, writing—review and editing. GMM: Conceptualization, supervision, methodology, project administration, writing—original draft, writing—review and editing. SJ: Conceptualization, supervision, methodology, project administration, writing—original draft, writing—review and editing. JC: Conceptualization, supervision, methodology, project administration, writing—original draft, writing—review and editing. SFJ: Conceptualization, methodology, resources, data curation, writing—original draft, writing—review and editing. DS: Resources, data curation, conceptualization, methodology, writing—original draft, writing—review and editing. SF: Resources, data curation, writing—original draft, writing—review and editing.
Additional information
Esteban Rodrigo Imedio, Ganesh M. Mugundu, and Juliann Chmielecki are no longer employees of AstraZeneca.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bauer, T.M., Moore, K.N., Rader, J.S. et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Targ Oncol 18, 517–530 (2023). https://doi.org/10.1007/s11523-023-00965-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-00965-7