
Abstract
Purpose
The purpose of this study is to assess treatment responses induced by the two tyrosine kinase inhibitors, Imatinib and Sunitinib, in a gastrointestinal stromal tumor (GIST) xenograft using a clinical positron emission tomography/computed tomography (PET/CT) scanner.
Methods
Nude mice bearing human GIST xenografts with mutations in exons 11 and 17 were randomly allocated to treatment with Imatinib, Sunitinib, or placebo daily for seven consecutive days. 2-deoxy-2-[18F]fluoro-d-glucose PET (18F-FDG-PET/CT) was performed in a clinical PET/CT scanner at baseline (day 0) and 1 and 7 days after onset of treatment. Treatment response was assessed by measuring tumor volumes and by calculation of tumor-to-liver 18F-FDG uptake ratios.
Results
Minor reductions in tumor volume were observed in both treatment groups. For the two treatment groups, significantly decreased tumor-to-liver uptake ratios were observed both at day 1 (Imatinib, −41%, p = .002; Sunitinib, −55%, p < .001) and at day 8 (Imatinib, −35%, p < .001; Sunitinib, −50%, p < .001), when compared to individual baseline values. For the control tumors, neither tumor volumes nor tumor-to-liver uptake ratios were altered during the 8 days the experiment lasted.
Conclusions
Significant anti-tumor effects were demonstrated following treatment with both Imatinib and Sunitinib. Decreased tumor-to-liver uptake ratios were more pronounced than tumor volume reductions. Effects of novel targeted therapies can be evaluated in the GIST xenograft model using a clinical PET/CT scanner.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal cancer of the digestive system and may occur in the entire length of the gastrointestinal tract [1]. GISTs develop from interstitial cells of Cajal [2] and typically express mutations in the c-KIT oncogene [3]. With an understanding of the key role of the KIT tyrosine kinase expression [4] and the subsequent introduction of the tyrosine kinase inhibitor (TKI) Imatinib, a new era in the management of this tumor entity began [5]. Imatinib has shown an eminent effect in patients with metastatic GIST [6], albeit often temporarily [7, 8]. Further understanding of the molecular basis of GIST oncogenesis has led to the development of a variety of novel agents for the treatment of Imatinib refractory GIST [9, 10], such as the multitargeted TKI, Sunitinib [11–13].
Computed tomography (CT) has conventionally been the method of choice to monitor patients with GIST. However, new targeted therapies for GIST have revealed insufficiency in the current solid tumor size-based response assessment criteria (RECIST), since initial treatment effect seldom is observed as tumor volume reduction [14, 15]. Recent studies have used decreased both in tumor size and in density values on CT for the evaluation of GIST response to Imatinib treatment [16], often referred to as the Choi criteria [17]. Treatment response is defined as more than 15% decrease in CT density value or more than 10% reduction in tumor size. Using the Choi criteria, a sensitivity of 97% and a specificity of 100% in identifying positron emission tomography (PET) responders have been reported [17]. In contrast, the corresponding values based on RECIST were 52% and 100%.
Most GISTs show high uptake of 2-deoxy-2-[18F]fluoro-d-glucose (18F-FDG) when imaged by PET, and treatment response can be observed early [6], even 24 h after onset of treatment [18]. Albeit still a controversial issue, a certain percentage of GISTs may probably be without any uptake of 18F-FDG and still be overtly malignant [16, 19].
As with Imatinib, KIT mutation status seems to be a predictor of response to Sunitinib [20, 21]. Knowledge on the relative responsiveness of different molecular subgroups of Imatinib-resistant GIST may optimize the treatment and thereby contribute to a better understanding of the mechanisms of resistance and how to circumvent them. The interest in other signal transduction inhibitors to overcome this resistance is thus growing.
The development of novel targeted therapies necessitates animal models, providing the same inherent molecular tumor heterogeneity as observed in the clinical setting in order to allow evaluation of treatment efficacy. To evaluate response to targeted therapies, it is important to assess the inhibition of tumor metabolism and not only tumor shrinkage. This stresses the importance of integrating molecular imaging technologies in treatment response evaluation.
The availability of dedicated small animal PET scanners is limited, and thus researchers have tried using adapted clinical systems [22–24]. Recently, Aide et al. demonstrated that a state-of-the art clinical PET/CT could provide high quality images and accurate quantification of the 18F-FDG uptake with comparable spatial resolution as dedicated small animal scanners [25]. Based on these findings, we conducted a preclinical therapy study assessing therapeutic efficacy in human GIST xenografts receiving Imatinib or Sunitinib treatment in a clinical PET/CT. This approach may, if rigorously validated, be used for high throughput evaluation of new TKIs and other pharmaceuticals being developed to circumvent treatment resistance.
The aims of this study were twofold: first, to evaluate treatment response to Imatinib or Sunitinib in the human GIST AHAX xenograft and second, to evaluate if a clinical PET/CT scanner can be used for this assessment.
Materials and Methods
Xenografts and Animals
The human GIST xenograft AHAX with mutations in exon 11 (c.1673_1687del, p.Lys558_Glu562del) and exon 17 (c.2446G > C, p.Asp816His) was used [26]. Xenografts were established by subcutaneous implantation of tumor tissue fragments (~2 × 2 × 2 mm3) bilaterally into 36 female NCR athymic mice (5–7 weeks; 25–30 g). The mice were bred at the animal department of our institution and kept under specific pathogen-free conditions at constant temperature (22–24°C) and humidity (55–60%). They were given sterilized food and tap water ad libitum. The animal study was approved by the Institutional Committee on Research Animal Care. All mice were ear-tagged and followed individually throughout the study. After 3 months, tumor volumes were 515 ± 45 mm3. At this time point, the 33 mice were randomly allocated into three groups, and baseline measurements were obtained. Three separate mice, not included in the treatment study, were used for comparing tumor-to-liver uptake ratio in a clinical and a dedicated animal PET scanner.
Treatment
Imatinib (Glivec®, Novartis Pharma GmbH, Basel, Switzerland) and Sunitinib (Sutent®, Pfizer Inc., New York, NY, USA) were pulverized and dissolved in distilled water or dissolved in 50 mM citrate buffer (pH = 3.5), respectively. Mice in the two treatment groups were given either 100 mg/kg Imatinib (group 1) or 40 mg/kg Sunitinib (group 2) by oral gavage once daily, whereas the control group received citrate buffer only (group 3). At the end of the experiment, the mice were sacrificed by neck dislocation.
Tumor Measurements
Tumor volumes were estimated by caliper measurements from implantation until the end of the experiment using the modified ellipsoid formulae [27, 28]: \( V\left( {{\hbox{m}}{{\hbox{m}}^3}} \right) = \left( {\pi /6} \right) \times a\left( {\hbox{mm}} \right) \times {b^2}\left( {{\hbox{m}}{{\hbox{m}}^2}} \right) \), with a and b being the longest and the perpendicular tumor diameters, respectively. Measurements were normalized to individual pre-treatment (day 0) tumor volumes.
Histopathological Examination
Material from the primary tumor and subsequent surgical specimens were reviewed by an experienced sarcoma pathologist. Paraffin-embedded tissue was processed for staining with hematoxylin and eosin (HE) and microscopic examination. The mitotic index was counted in ten high power fields (HPF; objective ×40, area of a single HPF; 0.3066 mm2).
18F-FDG PET/CT in a Clinical Scanner
PET examinations were performed before treatment (day 0), 24 h (day 1), and 7 days after onset of treatment with Imatinib, Sunitinib, or placebo. The mice were examined after 4 h of fasting using a clinical PET/CT scanner (Biograph 16, Siemens, Erlangen, Germany). The mice were anesthetized with 0.025 ml/10 g s.c. injections of tiletamine 2.4 mg/ml and zolazepam 2.4 mg/ml (Zoletil vet®, Virbac Laboratories, Carros, France), xylazine 3.8 mg/ml (Narcoxyl vet, Roche, Basel, Switzerland), and butorphanol 0.1 mg/ml (Torbugesic, Fort Dodge Laboratories, Fort Dodge, IA, USA) before examination. One hour after intraperitoneal injections of 7–10 MBq 18F-FDG (GE Healthcare AS, Oslo, Norway), the mice were positioned in groups side by side on a heating cushion. A CT scan was obtained with a slice thickness of 1 mm and a pixel size of 0.49 × 0.49 mm2. Subsequently, a 10-min one-bed position PET acquisition was obtained. Images were reconstructed by an OSEM iterative technique, using a 2-mm Gaussian post-reconstruction smoothing filter. The image format was 256 × 256, the pixel size was 2.67 × 2.67 mm2, and the slice thickness was 2 mm. Attenuation and scatter correction were applied before the images were transferred to a remote workstation for further image analysis.
18F-FDG PET in a Dedicated Animal Scanner
To validate the use of a clinical scanner for assessment of quantitative 18F-FDG uptake, three separate, untreated mice were subjected to 18F-FDG-PET examination in a dedicated small animal PET scanner (microPET Focus 120, Siemens Medical Solutions, Erlangen, Germany). Following 4 h of fasting, the mice were anesthetized, and 7–10 MBq 18F-FDG was injected intraperitoneally [29]. After 1 h, the mice were scanned for 10 min. Attenuation correction was obtained by a 10-min transmission scan with a 68Ge point source after 18F-FDG-PET. Data collected in list mode were reconstructed using 3-D OSEM-MAP [30–32] (2 OSEM iterations, 18 MAP iterations, β = 0.5, 128 × 128 × 95 matrix size, 0.87 × 0.87 × 0.80 mm3 voxel size). Images were transferred to a remote workstation for further analysis.
Image Analysis
The PET/CT images were analyzed using IDL (Interactive Data Language v6.2, Research Systems Inc., Boulder, CO, USA). Initially, an image volume covering all animals was reconstructed. All coronal planes were added to form a two-dimensional representation of the three-dimensional volume elements. These images were used to draw individual regions of interests (ROIs) around tumor and liver. The co-registered CT images were used for guiding the delineations. Tumor-to-liver uptake ratios for individual tumors were obtained by dividing mean tumor 18F-FDG uptake by the mean liver 18F-FDG uptake. All ratios were normalized to individual pre-treatment values.
Ex Vivo Counting
Immediately following clinical PET/CT, the three mice that underwent small animal PET examination 24 h earlier were sacrificed, and tumor and liver were harvested. Tissue samples were separately weighed and counted for 1 min in a gamma counter (Cobra II auto-gamma detector, Packard Instrument Company, Meriden, CT, USA). The samples were normalized to sample weight. Tumor-to-liver uptake ratios obtained from the clinical PET/CT scanner, the dedicated small animal PET scanner, and from ex vivo counting were compared.
Statistical Analysis
Statistical analyses were performed using SPSS version 16.0 (SPSS Science, Chicago, IL, USA). The Kolmogorov–Smirnov test was applied to test for normality. Statistical differences between groups were analyzed using one-way ANOVA, two-sided t test, and Pearson correlation coefficient. Significance was assigned at p ≤ .05.
Results
Tumor Growth
The 33 mice developed 49 tumors giving a tumor take of 74.2%. Prior to therapy, tumor volumes did not differ significantly between the three groups (p = .54). Initial tumor volumes were normally distributed (p = .19), and results are presented as means and standard deviations. Normalized tumor volumes for the two treatment groups and the control group as function of time after treatment are shown in Fig. 1. Tumor volume in the control group increased, although not significantly, with 11.5% from days 0 to 8 (p = .27). Compared to individual baseline values, the mean volume of the tumors in the Imatinib group was reduced by 15.7% at day 8 (p < .01). In the Sunitinib group, no statistically significant tumor decrease was observed (−8.8%, p = .41).

Normalized tumor volumes for the human GIST AHAX xenografts prior to therapy and after 1 and 7 days of daily Sunitinib, Imatinib, or placebo treatment. Error bars represent standard deviations.
The mitotic index in tissue samples obtained from all three groups was Imatinib, 2/10 HPF; Sunitinib, 8/10 HPF; and control, 21/10 HPF.
Treatment Response Assessed with PET/CT
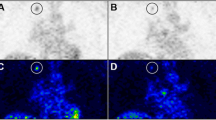
Mean number of pixels within the ROI used for the measurement of 18F-FDG uptake was 6.6 (range, 2–15) and 12.4 (range, 6–22) pixels for tumor and liver, respectively. Normalized tumor-to-liver ratio as function of time after onset of treatment is shown in Fig. 2. Pre-treatment metabolic activity was not significantly different between control and the two treatment groups (p = .358). One-way ANOVA revealed no changes in tumor-to-liver uptake ratios for the control group during the 8 days the experiment lasted (p = .536). One day after onset of therapy, normalized tumor-to-liver uptake ratios in the Imatinib- and Sunitinib-treated groups decreased by 40.8 ± 8.8% (p < .01) and 54.9 ± 8.1% (p < .01), respectively. At day 8, normalized tumor-to-liver uptake ratios were reduced by 35.2% (p < .001) and 50.1% (p < .001) for the Imatinib and the Sunitinib groups, respectively, relative to individual baseline values. It should, however, be noted that tumor-to-liver uptake ratios at days 1 and 8 were not significantly different (Imatinib, p = .56; Sunitinib, p = .64). An example of 18F-FDG uptake in a nude mouse prior to therapy and 24 h after start of Sunitinib treatment is shown in Fig. 3.

Normalized tumor-to-liver uptake ratios for the GIST AHAX xenografts prior to therapy and after 1 and 7 days of daily Imatinib, Sunitinib, or placebo treatment. Error bars represent standard deviations.

Clinical PET/CT images acquired before and 24 h after treatment with Sunitinib.
In Vivo Imaging Versus Ex Vivo Counting
Pearson correlation coefficient between tumor-to-liver obtained from ex vivo counting and from in vivo imaging was 0.99 for both the clinical PET/CT scanner and for the dedicated small animal PET scanner. It should however be noted that the absolute tumor-to-liver ratios were different; the clinical scanner yielded about 18% higher values, and the dedicated animal scanner yielded about 23% lower values, when compared to the ex vivo counting.
Discussion
This preclinical study demonstrates that tumor-to-liver 18F-FDG uptake ratio decreases following Imatinib and Sunitinib treatment in the human GIST AHAX xenograft, originating from a clinical Imatinib-resistant patients [26]. This study also shows that a clinical PET/CT can be used to assess treatment response in small animals.
For both treatment groups, significantly reduced 18F-FDG uptake was observed in the treated GIST xenografts 24 h after onset of treatment, and the level remained low during the entire experiment. The immediate metabolic shut-down has previously been reported for human GISTs [18]. This is in accordance with current knowledge that TKIs like Imatinib, directly targeting glucose uptake metabolism [33], will cause a rapid reduction in 18F-FDG uptake (within hours to days). The prevailing hypothesis is that the decrease reflects changes in glucose uptake mechanisms. Such early decrease in 18F-FDG uptake correlates to therapeutic outcome [34, 35], supporting its use as an early prognostic marker.
The AHAX xenograft originates from a patient with a clinically Imatinib-resistant GIST. The pronounced treatment effect of both Imatinib and Sunitinib is intriguing since the patient harbored mutations in exon 11 and exon 17, leading to treatment resistance. Loss of the genetic mutation leading to resistance could explain the observed treatment effect. Mutation analysis of the implanted xenograft tissue was however performed and revealed that both mutations (exon 11 and 17), as seen in the patient, were maintained in the xenografts [26].
Clinical experience has shown that tumors with mutations in exon 11 are less sensitive to Sunitinib treatment, and that exon 17 mutations (which encode the activation loop) are resistant to Imatinib [20] and have weak or no sensitivity to Sunitinib [36]. The observed response to both treatments suggests that AHAX xenograft contains both Imatinib- and Sunitinib-sensitive clones. This fits well with knowledge of the heterogeneity of GISTs, with numerous different mutations being harbored within a single tumor [37]. The maintenance of such heterogeneity after eight generations, which was the source of our implanted tumor material, may be due to a selection of the most virulent clones (or the clones harboring the highest mitotic rate). Imatinib resistance may also be incomplete. Low doses of either Imatinib or Sunitinib have been shown to have a partial inhibitory effect on KIT phosphorylation in a GIST cell line with mutations in exon 11 and exon 17, presumably due to inhibition of a minor population of cells with exon 11 mutations [36]. The antiangiogenic effects of Sunitinib [38] may also contribute to the observed treatment response.
In our study, the 18F-FDG uptake in the two treatment groups remained reduced for the 8 days the experiment lasted. For both groups, tumor-to-liver ratios increased slightly, but not significantly. This could possibly be attributed to the development of a selection pressure, promoting the growth of resistant clones or a change in glucose uptake mechanisms [39, 40].
The clinical PET/CT scanner has the advantage of being able to handle a large number of small animals simultaneously, thereby allowing high throughput imaging. Fig. 4 shows the experimental set-up in our study with ten mice within one bed position. Identical hardware, software, and acquisition protocols for examination of animals and humans ensure consistency of data and provide easy translation of developments from mouse to human in a clinical setting.

Representative CT (a), PET (b), and PET/CT (c) images of ten mice bearing AHAX xenografts obtained using a clinical PET/CT scanner. The anesthetized mice were positioned side by side on a heating cushion during acquisition of data.
Tumor-to-liver uptake ratios obtained from ex vivo counting differed in a systematic way from the ratios obtained from the clinical and the animal scanner. Because of intensity, diffusion signal intensities in small and irregular-shaped organs can never be accurately reproduced [41], and this effect is much more pronounced on a clinical PET scanner. The spatial resolution of the clinical PET/CT scanner is clearly inferior to that of the dedicated small animal PET scanner. With the actual Gaussian filter, the full-width at half maximum of the point spread function on the clinical scanner is 4.8 mm in the center of the field. The resulting point spread function smearing is most pronounced for small high-intensity objects. Systematic over- and underestimation of tumor-to-liver uptake ratios for the PET systems seem to have resulted from the way the ROIs were drawn at the different modalities. In order to obtain the most accurate assessment of tumor signal intensity in the clinical scanner images, a smaller ROI than the actual size of the tumor was used. On the other hand, CT information was used to draw the liver region, and the imaging process had also led to loss of intensity across the region border. Since the ratios were calculated on a per-average-pixel-value basis, this methodology is the probable reason for overestimation of the tumor-to-liver ratio from the measurements on the clinical scanner. The lack of co-registered CT images from the animal PET scanner may have led to the inclusion of signal intensities from tissues adjacent to or overlapping with the liver, resulting in a systematic increased average liver signal intensity and thus an underestimation of the tumor-to-liver ratio.
Despite the fact that the absolute ratios were different, the correlations between ratios obtained with different methods were very strong. Thus, the ratios obtained in this study are well suited for comparisons between groups, but less suitable for absolute quantification purposes. However, the present study demonstrated that the clinical PET/CT system yielded sufficient spatial resolution to obtain clinically relevant information from this animal model.
More accurate assessment of treatment response can be achieved by measuring tumor uptake as function of injected dose for individual mice. This approach, which we will use in our future studies, allows each animal to be its own control and will reduce the influence of intensity diffusion by allowing larger tumor ROIs to be drawn.
Conclusion
Significant anti-tumor effects of Imatinib and Sunitinib were demonstrated in a human GIST xenograft originating from a patient with a clinically Imatinib-resistant tumor. The measured treatment response, combined with the results from comparison between PET-assessed tumor-to-liver uptake ratio and ex vivo counting, demonstrates that a clinical PET/CT offers non-invasive and longitudinal assessment of treatment response to novel targeted therapies in preclinical models.
References
Miettinen M, Lasota J (2003) Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol 54(1):3–24
Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM (1998) Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 152(5):1259–1269
Corless CL, Fletcher JA, Heinrich MC (2004) Biology of gastrointestinal stromal tumors. J Clin Oncol 22(18):3813–3825
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S et al (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279(5350):577–580
Cassier PA, Blay JY (2010) Imatinib mesylate for the treatment of gastrointestinal stromal tumor. Expert Rev Anticancer Ther 10(5):623–634
Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D et al (2001) Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 344(14):1052–1056
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS et al (2008) Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 26(4):626–632
Shinomura Y, Kinoshita K, Tsutsui S, Hirota S (2005) Pathophysiology, diagnosis, and treatment of gastrointestinal stromal tumors. J Gastroenterol 40(8):775–780
Novak C, Trevino JG (2010) Gastrointestinal stromal tumors: a paradigm for therapeutic options in solid organ tumors. Mini Rev Med Chem 10(7):624–634
Reichardt P (2010) Optimal use of targeted agents for advanced gastrointestinal stromal tumours. Oncology 78(2):130–140
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G et al (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9(1):327–337
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW et al (2003) SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101(9):3597–3605
Demetri GD, Heinrich MC, Fletcher JA, Fletcher CD, Van den Abbeele AD, Corless CL et al (2009) Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res 15(18):5902–5909
Benjamin RS, Choi H, Macapinlac HA, Burgess MA, Patel SR, Chen LL et al (2007) We should desist using RECIST, at least in GIST. J Clin Oncol 25(13):1760–1764
Choi H (2008) Response evaluation of gastrointestinal stromal tumors. Oncologist 13(Suppl 2):4–7
Choi H, Charnsangavej C, de Castro FS, Tamm EP, Benjamin RS, Johnson MM et al (2004) CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol 183(6):1619–1628
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR et al (2007) Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 25(13):1753–1759
Shinto A, Nair N, Dutt A, Baghel NS (2008) Early response assessment in gastrointestinal stromal tumors with FDG PET scan 24 hours after a single dose of imatinib. Clin Nucl Med 33(7):486–487
Goerres GW, Stupp R, Barghouth G, Hany TF, Pestalozzi B, Dizendorf E et al (2005) The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylate. Eur J Nucl Med Mol Imaging 32(2):153–162
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J et al (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368(9544):1329–1338
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ et al (2006) Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24(29):4764–4774
Wolf G, Abolmaali N (2009) Imaging tumour-bearing animals using clinical scanners. Int J Radiat Biol 85(9):752–762
Robinson MK, Doss M, Shaller C, Narayanan D, Marks JD, Adler LP et al (2005) Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res 65(4):1471–1478
Seemann MD (2004) Human PET/CT scanners: feasibility for oncological in vivo imaging in mice. Eur J Med Res 9(10):468–472
Aide N, Desmonts C, Briand M, Meryet-Figuiere M, Poulain L (2010) High-throughput small animal PET imaging in cancer research: evaluation of the capability of the Inveon scanner to image four mice simultaneously. Nucl Med Commun 31(10):851–858
Revheim ME, Seierstad T, Berner JM, Bruland OS, Roe K, Ohnstad HO et al (2009) Establishment and characterization of a human gastrointestinal stromal tumour (GIST) xenograft in athymic nude mice. Anticancer Res 29(11):4331–4336
Euhus DM, Hudd C, LaRegina MC, Johnson FE (1986) Tumor measurement in the nude mouse. J Surg Oncol 31(4):229–234
Tomayko MM, Reynolds CP (1989) Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 24(3):148–154
Fueger BJ, Czernin J, Hildebrandt I, Tran C, Halpern BS, Stout D et al (2006) Impact of animal handling on the results of 18 F-FDG PET studies in mice. J Nucl Med 47(6):999–1006
Leahy R, Byrne C (2000) Recent developments in iterative image reconstruction for PET and SPECT. IEEE Trans Med Imaging 19(4):257–260
Qi J, Leahy RM, Cherry SR, Chatziioannou A, Farquhar TH (1998) High-resolution 3D Bayesian image reconstruction using the microPET small-animal scanner. Phys Med Biol 43(4):1001–1013
Qi J, Leahy RM (2000) Resolution and noise properties of MAP reconstruction for fully 3-D PET. IEEE Trans Med Imaging 19(5):493–506
Tarn C, Skorobogatko YV, Taguchi T, Eisenberg B, von Mehren M, Godwin AK (2006) Therapeutic effect of imatinib in gastrointestinal stromal tumors: AKT signaling dependent and independent mechanisms. Cancer Res 66(10):5477–5486
Holdsworth CH, Badawi RD, Manola JB, Kijewski MF, Israel DA, Demetri GD, Van den Abbeele AD (2007) CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol 189(6):W324–W330
Shankar LK, Hoffman JM, Bacharach S, Graham MM, Karp J, Lammertsma AA et al (2006) Consensus recommendations for the use of 18 F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute Trials. J Nucl Med 47(6):1059–1066
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D et al (2008) Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 26(33):5352–5359
Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC et al (2008) Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 216(1):64–74
Mena AC, Pulido EG, Guillen-Ponce C (2010) Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs 21(Suppl 1):S3–S11
Prenen H, Deroose C, Vermaelen P, Sciot R, biec-Rychter M, Stroobants S et al (2006) Establishment of a mouse gastrointestinal stromal tumour model and evaluation of response to imatinib by small animal positron emission tomography. Anticancer Res 26(2A):1247–1252
Wardelmann E, Thomas N, Merkelbach-Bruse S, Pauls K, Speidel N, Buttner R et al (2005) Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol 6(4):249–251
Skretting A (2009) Intensity diffusion ‘is a better description than’ partial volume effect. Eur J Nucl Med Mol Imaging 36(3):536–537
Acknowledgements
The authors greatly acknowledge the technical assistance provided by chief technologist Kari Bjering, consultant pathologist Bodil Bjerkehagen, chief engineer Hong Qu, nuclear medicine technologist Inger Hagen, and department engineer Alexandr Kristian. This work was financially supported by the South-Eastern Norway Regional Health Authority grant 2009070 (KR), the Norwegian Cancer Society grant 80114001 (TS), and Novartis for an unrestricted research grant (MER).
Conflicts of Interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Revheim, ME., Røe, K., Bruland, Ø.S. et al. Monitoring the Effect of Targeted Therapies in a Gastrointestinal Stromal Tumor Xenograft Using a Clinical PET/CT. Mol Imaging Biol 13, 1234–1240 (2011). https://doi.org/10.1007/s11307-010-0464-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-010-0464-0