
Abstract
There is sparse information about specific storage and handling protocols that minimize analytical error and variability in samples evaluated by targeted metabolomics. Variance components that affect quantitative lipid analysis in a set of human serum samples were determined. The effects of freeze-thaw, extraction state, storage temperature, and freeze-thaw prior to density-based lipoprotein fractionation were quantified. The quantification of high abundance metabolites, representing the biologically relevant lipid species in humans, was highly repeatable (with coefficients of variation as low as 0.01 and 0.02) and largely unaffected by 1–3 freeze-thaw cycles (with 0–8% of metabolites affected in each lipid class). Extraction state had effects on total lipid class amounts, including decreased diacylglycerol and increased phosphatidylethanolamine in thawed compared with frozen samples. The effects of storage temperature over 1 week were minimal, with 0–4% of metabolites affected by storage at 4°C, −20°C, or −80°C in most lipid classes, and 19% of metabolites in diacylglycerol affected by storage at −20°C. Freezing prior to lipoprotein fractionation by density ultracentrifugation decreased HDL free cholesterol by 37% and VLDL free fatty acid by 36%, and increased LDL cholesterol ester by 35% compared with fresh samples. These findings suggest that density-based fractionation should preferably be undertaken in fresh serum samples because up to 37% variability in HDL and LDL cholesterol could result from a single freeze-thaw cycle. Conversely, quantitative lipid analysis within unfractionated serum is minimally affected even with repeated freeze-thaw cycles.
Similar content being viewed by others


1 Introduction
Improper storage and handling of serum samples prior to compositional analysis by targeted metabolomics and lipoprotein separation may significantly influence accuracy compared with the analysis of fresh samples. Yet there is little information about specific storage conditions and handling protocols that can minimize error and variability. The only literature available examines the effects of sample storage and handling on the enzymatic analysis of total cholesterol (TC) and triacylglycerol (TG). The results of these experiments have been mixed. In some studies, freezing and storage time increased lipid concentrations (Evans et al. 1997; Pini et al. 1990; Tiedink and Katan 1989; Wood et al. 1980); in other studies, lipid concentrations decreased (Bausserman et al. 1994; Donnelly et al. 1995; Ekbom et al. 1996; Evans et al. 1995, 1997; Nanjee and Miller 1990; Simo et al. 2001; Tiedink and Katan 1989); and other studies found either increases or decreases depending on initial concentrations (Bachorik et al. 1980, 1982). Still other investigators found no significant changes in lipid concentration (Bausserman et al. 1994; Donnelly et al. 1995; Kuchmak et al. 1982; Nanjee and Miller 1990; Stokes et al. 1986; Wood et al. 1980). Only two studies examined the effects of multiple freeze-thaw cycles. Samples that were stored for 24 months at −80°C and that had undergone 1–3 freeze-thaw cycles showed compositional changes after 24 months as analyzed with enzymatic detection kits (Kronenberg et al. 1994), but there were no differences between 1 and 3 freeze-thaw cycles. Another study also found no differences, but freeze-thawed samples were compared only with each other and not with fresh samples (Comstock et al. 2001).
To our knowledge there is no information regarding the storage and handling of serum samples and their effects on the composition of fatty acids (FA) within separated lipid classes as measured by quantitative lipid analysis, within whole serum or density fractionated serum. Therefore, the aims of this study were to examine in detail the storage and handling conditions that influence the quality of lipid compositional analyses using a combination of platforms, including preparative HPLC and TLC, and gas chromatography (GC)-flame ionization detection (FID). In particular, we sought to determine the effects of multiple freeze-thaw cycles, the state of the sample (frozen or thawed) at the time of extraction, storage temperature, and freezing serum prior to lipoprotein fractionation on quantitative lipid analysis in human samples.
2 Materials and methods
Two experiments were performed to investigate the overall effects of sample storage and handling. For Experiment 1, samples were purchased from PromedDx (Norton, MA), and for Experiment 2, fresh serum samples were collected from a healthy volunteer after an overnight fast. Methods, experimental design, and statistical analyses specific to each experiment are described below.
2.1 Quantitative lipid analysis
In both Experiment 1 and Experiment 2, quantitative lipid analysis was performed by Lipomics Technologies, Inc. (West Sacramento, CA) according to the method of Watkins et al. (2002). Briefly, lipids were extracted in the presence of authentic internal standards by the method of Folch et al. (1957) using chloroform-methanol (2:1, v/v) + 0.05% butylated hydroxytoluene. Serum (200 μl) was used for each analysis. Individual lipid classes—TG, diacylglycerol (DG), free fatty acids (FFA), phosphatidylcholine (PC), lysophosphatidylcholine (LY), phosphatidylethanolamine (PE), cholesterol ester (CE), and free cholesterol (FC)—within each extract were separated by preparative HPLC (Cao et al. 2008) for the phospholipid classes and by thin-layer chromatography for the neutral lipid classes. Each isolated lipid class fraction (except FC, as this lipid class contains no FA) was trans-esterified in 3 N methanolic-HCl in a sealed vial under N2 at 100°C for 60 min. The resulting FA methyl esters were extracted from the mixture with hexane containing 0.05% butylated hydroxytoluene and prepared for GC by sealing the hexane extracts under N2. FA methyl esters were separated and quantified by capillary GC using a gas chromatograph (Agilent Technologies model 6890, Wilmington, DE) equipped with a 30-m DB-225MS capillary column (Agilent Technologies, Folsom, CA) and a flame-ionization detector. A total of 37 FA were measured in each lipid class and included the following: 14:0, 15:0, 16:0, 18:0, 20:0, 22:0, 24:0, 14:1n5, 16:1n7, trans (t)16:1n7, 18:1n9, t18:1n9, 18:1n7, 18:2n6, t18:2n6, 18:3n6, 18:3n3, 18:4n3, 20:1n9, 20:2n6, 20:3n9, 20:3n6, 20:4n6, 20:3n3, 20:4n3, 20:5n3, 22:1n9, 22:2n6, 22:4n6, 22:5n3, 22:6n3, 24:1n9, and 24:6n3, and the plasmalogen derivatives of 16:0, 18:0, 18:1n9, and 18:1n7.
2.2 Data processing and statistics
The determined amounts of individual FA were used to calculate the mass of individual lipid classes, which were expressed as nmol FA/g serum. Individual FA data were analyzed as mol.% calculated as the percentage of each individual FA relative to the sum of the concentrations of all FA within that lipid class. Lipid class composition was analyzed as both individual FA concentrations and as categories of FA: SFA, saturated; MUFA, monounsaturated; PUFA, polyunsaturated; and n3, n6, n7, and n9: omega-3, -6, -7, and -9, respectively. The data were pre-processed before formal statistical methods were applied in order to account for the large data density of metabolomic analyses. Metabolites with >30% missing observations were excluded, and observations that were >4 standard deviations from the mean were excluded for statistical analysis after lipid class totals were calculated.
Change detection plots were used to determine whether the observed signal was greater than that which could be expected by chance (noise). Change detection plots were produced in the following way: P-values for the appropriate comparison were calculated for each metabolite using a Student’s t-test and ranked from smallest to largest. The log of the rank versus the log of the P value for each comparison of interest was plotted. The distribution of P-values expected by chance at each rank was determined by a Monte Carlo permutation method, using values for each group that had been scaled to the mean and combined. The Nth ranked P-values from many permutations were used to construct the Nth boxplot. The y-axis value at which each rank versus log P-value line intercepts the vertical line indicates how many observed P-values exceeded the smallest P-value expected by chance.
Data were assessed for normality with histograms. If non-normality was detected, a log transformation was performed and the data were then re-analyzed. The results (P-values) of the log-transformed data were similar to the results of the non-transformed data, therefore the non-transformed data were used for analysis and reporting.
All statistics were performed using the software program R (The R Foundation for Statistical Computing).
2.3 Experiment 1: study design, methods, and statistics
The hypothesis was that multiple freeze-thaw cycles, pre-extraction state (frozen or thawed), and storage temperature lead to changes in FA concentrations within serum samples. The specific aims of this experiment were to determine the effects of these three key factors. (1) The effect of the number of freeze-thaw cycles was determined by comparing fresh samples with samples that had been frozen once, twice, or three times. (2) The effect of extraction state was determined by comparing samples that had been extracted frozen with those that had been extracted thawed. (3) The effect of storage temperature was determined by comparing samples that had been stored for 1 week at 4°C, −20°C, and −80°C. In order to address these specific aims, the following experimental protocol was performed.
Fresh, non-fasting serum samples from 3 healthy volunteers were purchased from PromedDx (Norton, MA). The blood was collected according to routine venipuncture protocols by a Food and Drug Administration-regulated facility, and serum was separated from blood by centrifugation according to routine protocols. The samples were refrigerated (i.e. never frozen), shipped the day after blood collection on cold packs, and analyzed within 48 h of collection, therefore, these samples were considered to be “fresh.”
Three batches of serum, each from a different individual, were tested under all conditions. Three replicates for each batch within each treatment type were tested, generating a total of nine samples for each analysis. Each group of samples was tested under the following conditions: (a) fresh, never frozen; (b) 1 freeze-thaw cycle; (c) 2 freeze-thaw cycles; (d) 3 freeze-thaw cycles; (e) frozen once, lipids extracted directly from frozen sample; (f) 1 freeze-thaw cycle, frozen again, lipids extracted directly from frozen sample; (g) stored at 4°C for 1 week; (h) 1 freeze-thaw cycle, stored at −20°C for 1 week; and (i) 1 freeze-thaw cycle, stored at −80°C for 1 week. Treatment (a) samples were extracted as soon as they arrived in the laboratory. Aliquots of all other samples were frozen as rapidly as possible. Samples from treatments (e) and (f) were extracted and analyzed at the same time. Treatments (g), (h), and (i) samples were extracted and analyzed at the same time.
Coefficients of variation (CV)’s were calculated as the standard deviation divided by the mean for each lipid metabolite measured (e.g. all FA within all lipid classes) for 5 sample replicates of a fresh serum sample from Experiment 1. The results are presented in a figure and illustrate the level of variability in the measurement of fresh samples. The effects of number of freeze-thaw cycles (1× , 2× , 3×) were examined with one-way ANOVA by comparing samples that were frozen once (treatment b), twice (treatment c), and three times (treatment d) to fresh samples (treatment a). Post-hoc analysis by t-test was employed to determine significant differences between each treatment (b, c, and d) and fresh samples (treatment a). The effects of extracting frozen compared with thawed samples (treatments b + c vs. e + f) were assessed by t-test. The temperature effect over 1 week of storage at 4°C, −20°C, and −80°C was compared using one-way ANOVA. Post-hoc analysis by t-test was used to determine significant differences between storage for 1 week at each temperature and fresh samples.
2.4 Experiment 2: study design, methods, and statistics
We hypothesized that freezing would affect lipid composition within lipoproteins separated by density gradient ultracentrifugation. A blood sample was collected from one healthy adult after an overnight fast. The Institutional Review Board of the University of California Davis approved the study protocol, and written informed consent was obtained from the subject. The blood was allowed to clot in the blood collection tube at room temperature for 30 min, and was then centrifuged at 1,600 rpm and 4°C for 10 min. The serum was divided into 6 2-ml aliquots within 60 min of blood collection. Three of these samples were kept fresh on ice (e.g. never frozen). The other three samples were promptly placed in a −80°C freezer, kept frozen for 2 h, and then thawed on ice for 30 min.
The samples were separated at the same time by sequential ultracentrifugation using a modification of the fast separation micromethod reported by Brousseau et al. (Brousseau et al. 1993). The centrifugation procedure was started within 6 h of blood collection and completed on the same day, using a Sorvall RC-120GX Micro-Ultracentrifuge (Sorvall S120-AT2 rotor, 120,000 rpm). To separate the VLDL, 600 μl of d = 1.006 kg/l NaCl solution was transferred to each tube, underlain with 1.2 mL of fresh or thawed sample, and submitted to ultracentrifugation (1 h 23 min, 8°C). The top 400 μl of sample was collected by aspiration as VLDL. To prepare LDL, the remaining sample was overlain with d = 1.34 kg/l NaCl/NaBr solution, submitted to ultracentrifugation (2 h 5 min, 8°C), and the top 400 μl collected by aspiration. The HDL were prepared similarly, by overlaying with d = 1.21 kg/l NaCl/NaBr solution, followed by ultracentrifugation (3 h 30 min, 8°C) and aspiration. All resulting fractions were immediately frozen and stored at −80°C until lipid compositional analysis was performed as described above.
Because there were only two factor levels (fresh and frozen), a two-sided unpaired t-test was used, with the assumption that the variances between the two groups were equal. P values from the unpaired t-test and Bartlett’s test for variance were calculated for each metabolite in each lipoprotein fraction separately. When the P-value of the t-test was <0.05, and the P value of the Bartlett test was >0.05, the mean metabolite concentration in the fresh vs. frozen sample after ultracentrifugation was significantly different.
3 Results
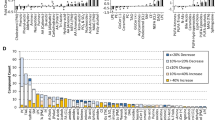
The level of variability in the measurement of all of the metabolites in 5 replicates of a fresh serum sample is shown in Fig. 1 as the CVs for each metabolite (e.g. each FA within each lipid class). The CVs were low for all of the high abundance metabolites, with values as low as 0.01 and 0.02 for many of the key metabolites of biological relevance in humans (e.g. TG18:1n9, TG total lipid class, CE16:0, TG16:0, FFA18:1n9, CE20:5n3, CE20:4n6). Seventy percent of the metabolites measured had CVs <0.33, 50% of the metabolites had CVs <0.15, and CVs for the total lipid classes were 0.07, 0.29, 0.08, 0.03, 0.13, 0.06, and 0.02 for CE, DG, FFA, LY, PC, PE, and TG, respectively.

Coefficients of variation (CV)s for metabolites measured in fresh serum. The CVs were calculated for each metabolite measured (e.g. each fatty acid within each lipid class) in 5 replicates of the same fresh serum sample from Experiment 1. The CVs were plotted in decreasing order for all metabolites. Low abundance metabolites (e.g. DG20:4n3, PE14:1n5) had higher CVs whereas high abundance metabolites (e.g. TG18:1n9, CE16:0) had very low CVs as low as 0.01 and 0.02. This illustrates the high reproducibility of the analytical method for metabolites of biological significance in humans
A summary of results for Experiment 1 is shown in Table 1, showing total amounts of each lipid class recovered from fresh serum, and the % of metabolites within each lipid class that was found to be significantly affected by each treatment. A similar summary of results for Experiment 2 is shown in Table 5, showing total amount of lipid and total amounts of each lipid class recovered in the unfractionated serum and within each lipoprotein fraction, as well as the % of metabolites within each lipid class for each lipoprotein class that was found to be significantly affected by freezing. The number of metabolites excluded from statistical analysis based on the stated criteria in Sect. 2.2 for each lipid class was 7, 11, 5, 6, 1, 2, and 4 in CE, DG, FFA, LY, PC, PE, and TG, respectively. These metabolites tended to be very low abundance metabolites including 22:2n6, 18:4n3, 20:3n3, 24:6n3, dm16:0, dm18:0, dm18:1n7, dm18:1n9, t16:1n7, t18:1n9, and t18:2n6.
There were surprisingly few changes in serum lipid composition that resulted from multiple freeze-thaw cycles in the present study. In Experiment 1, <1% of metabolites within each lipid class in samples that had undergone 1, 2, or 3 freeze-thaw cycles were significantly different from those in fresh samples. Post-hoc analyses were performed to determine specific changes between each treatment, and all comparisons were examined. Shown in Table 2 are only the metabolites (out of 262) that were significantly different from fresh. DG20:3n9, FFA22:5n3, PE18:0, and PE20:4n3 were significantly decreased after 1 freeze-thaw cycle. The reductions in DG20:3n9, FFA22:5n3, and PE20:4n3 were on the order of 50–90%, whereas the reduction in PE18:0 was only about 9% from fresh. CE20:2n6 was reduced 50–60% after two and three freeze-thaw cycles. DG20:3n9, DG20:2n6, LY20:5n3 were also decreased 30–35% after two freeze-thaw cycles, whereas LY16:0 and TG22:0 were increased 8% and 38%, respectively, after two freeze-thaw cycles.
The extraction of frozen samples compared with thawed samples had effects on whole lipid classes, specifically DG and PE, which were altered in opposite directions. As shown in Table 3, 9 out of 262 metabolites were significantly affected by extraction state, including increases in two dimethyl (dm) FA, PCdm18:0 and PCdm18:1n9, which increased about 40%, and an increase in PE of 27% when extracted thawed compared with frozen. On the other hand, DG18:3n6, LY20:4n6, LY22:5n3, PE16:0, and LYPUFA decreased 10–50%, and DG decreased 26% when extracted thawed compared with frozen.
The effects of storage temperature for 1 week were minor, with 0–4% of metabolites significantly affected at 4°C, −20°C, and −80°C in 7 out of 8 lipid classes (CE, FFA, LY, PC, PE, and TG) and 19% of metabolites affected by storage at −20°C in the DG lipid class (Table 1). Samples stored at 4°C had 20–50% lower concentrations of 5 DG metabolites and a 50% decrease in CE20:2n6 compared with fresh samples (Table 4). Samples stored at −20°C had 50% lower CE20:2n6 and DG20:3n9 but 45% higher TG22:0 than fresh samples, and samples stored at −80°C had 50% lower DGdm and 3% lower PCn7 species, whereas TG22:0 was again increased 45% compared with fresh.
In Experiment 2, paired Student’s t-tests were used to compare metabolites measured in fresh compared with frozen serum that was separated into VLDL, LDL, and HDL (Table 5). Within HDL, 10 out of 262 metabolites were affected by freezing prior to fractionation, including a 21% loss of PC18:2n6 and 37% loss of free cholesterol (FC) total lipid class, and increases in several LY and saturated PC species compared with fresh samples (Table 6). Within LDL from frozen samples, 13 out of 262 metabolites changed significantly relative to LDL from fresh samples, among them a 35% increase in CE, as well as increases across several long-chain TGPUFA (Table 7). Within VLDL, 15 out of 262 metabolites were significantly affected, including increases in several LY species, decreases in several TG species, and a significant 36% decrease in FFA total lipid class (Table 8).
4 Discussion
Variance components in processing and handling are additive, and can therefore skew results significantly. Whereas past studies examined the effects of storage and handling conditions on lipid composition measured with enzymatic kits and immuno-detection, in the current study, compositional analysis was performed using a platform combining preparative HPLC and TLC followed by quantitative GC-FID. Structural changes in lipoprotein particles and lipid microenvironments that occur during freeze-thaw cycles and storage at low temperatures likely impact the quality of enzymatic detection of lipid composition. However, there may be fewer impacts on lipid composition itself, as measured by GC-FID, as shown in this study. Less than 1% of metabolites across 8 classes of lipids were affected significantly by one, two, or three freeze-thaw cycles.
Storage for 1 week at 4°C, −20°C, and −80°C had only minor effects on serum lipid composition, with 0–4% of metabolites affected in most lipid classes. Longer storage durations, which were not examined in this study, may result in more differences. An earlier study found increasing effects of storage duration from 11 to 27 weeks (Tiedink and Katan 1989). Further studies are needed to examine the effects of temperature with storage durations longer than 1 week. Even at 1 week, storage at lower temperatures (−20°C and −80°C compared with 4°C) was beneficial and reduced alterations to the composition of DG species in particular, 19% of which were affected by storage at −20°C.
Freezing of samples prior to lipoprotein separation via density ultracentrifugation significantly changed HDL and LDL cholesterol concentrations as well as VLDL FFA concentrations. Structural changes in lipoproteins caused by freezing and thawing may have affected the density characteristics of particles and/or caused the aggregation of particles (Castile and Taylor 1999; Cevc and Richardsen 1999). Other possible causes for observed differences in metabolites resulting from freeze-thaw are enzymatic hydrolysis and synthesis, enzymatic transfer of lipids between lipoproteins, and non-enzymatic oxidation. The hydrolysis of phospholipids, TG, and DG by lipases, and CE by esterases remaining in the serum, which may be re-activated after each thawing, may have been involved. Increased hydrolysis product coupled with decreased substrate after freezing may be indicative of lipase activity. For example, in HDL LY18:2n6 increased, whereas PC18:2n6 decreased after freeze-thaw. Lecithin:cholesterol acyltransferase associated with HDL, and cholesterol ester transfer protein associated with HDL, VLDL, and LDL may have been responsible for the synthesis of new CE from FC and FFA, and the transfer of lipids between lipoproteins, respectively. There was a 37% decrease in HDL FC and a concomitant 36% decrease in VLDL FFA accompanied by a 35% increase in LDL CE in frozen compared with fresh serum. The non-enzymatic oxidation of certain more volatile species (e.g. longer-chain, more unsaturated FA) may also have been involved. For example, several long-chain PUFA species within VLDL TG, including TG22:4n6 and TG20:4n3, were reduced. However, these are also very low abundance metabolites, and thus the reason for the variability in fresh vs. frozen samples may have been due to the inherent variability in their measurement as low abundance metabolites rather than due to oxidation.
The findings of this study suggest that with careful and consistent handling, unfractionated serum samples can be stored for later quantitative lipid analysis with minor effects on quantitative lipid composition for most of the biologically relevant lipid species in humans. We suggest that the prompt storage of the smallest reasonable aliquot volume at the lowest possible temperature (e.g. −80°C) is preferable for reducing variability in the quantitative measurement of lipid metabolites introduced by freezing. If samples need to be thawed in order to divide already frozen samples into smaller aliquots for several different analyses/procedures, thawing samples on ice, followed by prompt redistribution of the larger sample into several smaller aliquots, and prompt refreezing until analysis should minimize any variability that could be introduced by the additional handling. It must be noted that no samples with visible crystallization, separation, or discoloration were included in this study, and therefore, the changes that could occur to lipid composition in samples that were collected or handled in such as way as to cause these observable changes were not tested.
On the other hand, density-based fractionation of lipoproteins after freezing was associated with significant changes to HDL and LDL cholesterol concentrations, and VLDL FFA along with relative increases in LDL TG metabolites and decreases in VLDL TG metabolites, which would suggest that the transfer of lipids, and/or physical changes to the structure of the lipoproteins occurred during freeze-thaw. Thus, density-based lipoprotein fractionation of frozen serum samples should be undertaken with caution.
One potential limitation of our study is that in Experiment 2, serum samples from only one donor were studied. We did not aim to address the question of inter-individual or biological variability among individuals, which is a topic of great interest in the clinical research community, and which has been studied previously (Ruhli et al. 2008; Zivkovic et al. 2008). Instead, to address the methodological question of how freeze-thaw affects sample stability with regard to quantitative lipid analysis following lipoprotein fractionation, we analyzed the serum sample in triplicate.
5 Concluding remarks
Important clinical samples often need to be stored prior to analysis. It is also often the case that additional analyses need to be performed on existing samples that represent a precious resource. Samples may be from especially vulnerable populations or may represent unique opportunities that would be difficult to replicate. In all of these situations, the proper storage and handling of samples is paramount to retaining their informativeness. The findings of this study suggest that if samples are handled with care, it is possible to effectively eliminate the potential variability introduced by handling and storage for quantitative lipid compositional analysis in unfractionated serum for the majority of metabolites that are of biological relevance in humans (e.g. the high abundance metabolites). However, freezing prior to density-based fractionation does introduce significant variability particularly in HDL and LDL cholesterol as well as LDL and VLDL TG concentrations.
Abbreviations
- TC:
-
Total cholesterol
- TG:
-
Triacylglycerol
- FA:
-
Fatty acid
- DG:
-
Diacylglycerol
- FFA:
-
Free fatty acid
- GC:
-
Gas chromatography
- PC:
-
Phosphatidylcholine
- LY:
-
Lysophosphatidylcholine
- PE:
-
Phosphatidylethanolamine
- CE:
-
Cholesterol ester
- SFA:
-
Saturated fatty acid
- MUFA:
-
Monounsaturated fatty acid
- PUFA:
-
Polyunsaturated fatty acid
- CV:
-
Coefficient of variation
- FC:
-
Free cholesterol
- dm:
-
Dimethyl
References
Bachorik, P. S., Walker, R., Brownell, K. D., Stunkard, A. J., & Kwiterovich, P. O. (1980). Determination of high density lipoprotein-cholesterol in stored human plasma. Journal of Lipid Research, 21(5), 608–616.
Bachorik, P. S., Walker, R. E., & Kwiterovich, P. O., Jr. (1982). Determination of high density lipoprotein-cholesterol in human plasma stored at −70 degrees C. Journal of Lipid Research, 23(8), 1236–1242.
Bausserman, L. L., Saritelli, A. L., & Milosavljevic, D. (1994). High-density lipoprotein subfractions measured in stored serum. Clinical Chemistry, 40(9), 1713–1716.
Brousseau, T., Clavey, V., Bard, J. M., & Fruchart, J. C. (1993). Sequential ultracentrifugation micromethod for separation of serum lipoproteins and assays of lipids, apolipoproteins, and lipoprotein particles. Clinical Chemistry, 39(6), 960–964.
Cao, H., Gerhold, K., Mayers, J. R., Wiest, M. M., Watkins, S. M., & Hotamisligil, G. S. (2008). Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell, 134(6), 933–944.
Castile, J. D., & Taylor, K. M. (1999). Factors affecting the size distribution of liposomes produced by freeze-thaw extrusion. International Journal of Pharmaceutics, 188(1), 87–95.
Cevc, G., & Richardsen, H. (1999). Lipid vesicles and membrane fusion. Advanced Drug Delivery Reviews, 38(3), 207–232.
Comstock, G. W., Burke, A. E., Norkus, E. P., Gordon, G. B., Hoffman, S. C., & Helzlsouer, K. J. (2001). Effects of repeated freeze-thaw cycles on concentrations of cholesterol, micronutrients, and hormones in human plasma and serum. Clinical Chemistry, 47(1), 139–142.
Donnelly, J. G., Soldin, S. J., Nealon, D. A., & Hicks, J. M. (1995). Stability of twenty-five analytes in human serum at 22 degrees C, 4 degrees C, and −20 degrees C. Pediatric Pathology and Laboratory Medicine, 15(6), 869–874.
Ekbom, T., Lindholm, L. H., Lanke, J., & Nilsson-Ehle, P. (1996). Decrease in high density lipoprotein cholesterol during prolonged storage. CELL Study Group. Scandinavian Journal of Clinical and Laboratory Investigation, 56(2), 97–101.
Evans, K., Mitcheson, J., & Laker, M. F. (1995). Effect of storage at 4 degrees C and −20 degrees C on lipid, lipoprotein, and apolipoprotein concentrations. Clinical Chemistry, 41(3), 392–396.
Evans, K., Mitcheson, J., & Laker, M. F. (1997). Effect of storage at −70 degrees C on lipid, lipoprotein and apolipoprotein concentrations. Clinica Chimica Acta, 258(2), 219–229.
Folch, J., Lees, M., & Sloane-Stanley, G. H. (1957). A simple method for the isolation and purification of total lipids from animal tissue. Journal of Biological Chemistry, 226, 497–509.
Kronenberg, F., Lobentanz, E. M., Konig, P., Utermann, G., & Dieplinger, H. (1994). Effect of sample storage on the measurement of lipoprotein[a], apolipoproteins B and A-IV, total and high density lipoprotein cholesterol and triglycerides. Journal of Lipid Research, 35(7), 1318–1328.
Kuchmak, M., Taylor, L., & Olansky, A. S. (1982). Suitability of frozen and lyophilized reference sera for cholesterol and triglyceride determinations. Clinica Chimica Acta, 120(2), 261–271.
Nanjee, M. N., & Miller, N. E. (1990). Evaluation of long-term frozen storage of plasma for measurement of high-density lipoprotein and its subfractions by precipitation. Clinical Chemistry, 36(5), 783–788.
Pini, C., Sommariva, D., Branchi, A., Scandiani, L., & Fasoli, A. (1990). Effects of serum storage on the determination of cholesterol. La Ricerca in clinica e in laboratorio, 20(1), 37–44.
Ruhli, F. J., Henneberg, M., Schaer, D. J., Imhof, A., Schleiffenbaum, B., & Woitek, U. (2008). Determinants of inter-individual cholesterol level variation in an unbiased young male sample. Swiss Medical Weekly, 138(19–20), 286–291.
Simo, J. M., Camps, J., Vilella, E., Gomez, F., Paul, A., & Joven, J. (2001). Instability of lipoprotein(a) in plasma stored at −70 degrees C: effects of concentration, apolipoprotein(a) genotype, and donor cardiovascular disease. Clinical Chemistry, 47(9), 1673–1678.
Stokes, Y. M., Salmond, C. E., Carpenter, L. M., & Welby, T. J. (1986). Stability of total cholesterol, high-density-lipoprotein cholesterol, and triglycerides in frozen sera. Clinical Chemistry, 32(6), 995–999.
Tiedink, H. G., & Katan, M. B. (1989). Variability in lipoprotein concentrations in serum after prolonged storage at −20 degrees C. Clinica Chimica Acta, 180(2), 147–155.
Watkins, S. M., Reifsnyder, P. R., Pan, H. J., German, J. B., & Leiter, E. H. (2002). Lipid metabolome-wide effects of the PPARgamma agonist rosiglitazone. Journal of Lipid Research, 43(11), 1809–1817.
Wood, P. D., Bachorik, P. S., Albers, J. J., Stewart, C. C., Winn, C., & Lippel, K. (1980). Effects of sample aging on total cholesterol values determined by the automated ferric chloride-sulfuric acid and Liebermann-Burchard procedures. Clinical Chemistry, 26(5), 592–597.
Zivkovic, A. M., Wiest, M. M., Nguyen, U., Nording, M. L., Watkins, S. M., & German, J. B. (2008). Assessing individual metabolic responsiveness to a lipid challenge using a targeted metabolomic approach. Metabolomics. doi:10.1007/s11306-008-0136-0.
Acknowledgements
The authors acknowledge Cora J. Dillard for her invaluable help with editing. This work was supported by University of California at Davis Graduate Group in Nutrition Block Grant, Jastro Shields Scholarship, and Superfund Training Fellowship to A. M. Zivkovic; supported in part by the National Institute of Environmental Health Sciences (NIEHS) grant R37 ES02710, the NIEHS Superfund Basic Research Program P42 ES04699, the University of California Davis Center for Children’s Environmental Health, NIEHS grant P01 ES11269, and the University of California Discovery Program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Zivkovic, A.M., Wiest, M.M., Nguyen, U.T. et al. Effects of sample handling and storage on quantitative lipid analysis in human serum. Metabolomics 5, 507–516 (2009). https://doi.org/10.1007/s11306-009-0174-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-009-0174-2