
Abstract
G protein-coupled receptors (GPCRs) are a major drug target and can be activated by a range of stimuli, from photons to proteins. Despite the progress made in the last decade in molecular and structural biology, their exact activation mechanism is still unknown. Here we describe new insights in specific regions essential in adenosine A2B receptor activation (A2BR), a typical class A GPCR. We applied unbiased random mutagenesis on the middle part of the human adenosine A2BR, consisting of transmembrane domains 4 and 5 (TM4 and TM5) linked by extracellular loop 2 (EL2), and subsequently screened in a medium-throughput manner for gain-of-function and constitutively active mutants. For that purpose, we used a genetically engineered yeast strain (Saccharomyces cerevisiae MMY24) with growth as a read-out parameter. From the random mutagenesis screen, 12 different mutant receptors were identified that form three distinct clusters; at the top of TM4, in a cysteine-rich region in EL2, and at the intracellular side of TM5. All mutant receptors show a vast increase in agonist potency and most also displayed a significant increase in constitutive activity. None of these residues are supposedly involved in ligand binding directly. As a consequence, it appears that disrupting the relatively “silent” configuration of the wild-type receptor in each of the three clusters readily causes spontaneous receptor activity.
Similar content being viewed by others


Introduction
The adenosine receptors form a small subfamily of class A G protein-coupled receptors (GPCRs). Four subtypes of adenosine receptors are known (A1 receptor (A1R), A2A receptor (A2AR), A2B receptor (A2BR), and A3 receptor (A3R)) that all bind the endogenous ligand adenosine. The A1R and the A3R subtypes are coupled to Gi proteins, hereby mediating the inhibition of adenylyl cyclase causing decreased levels of cAMP in the cell. The A2AR and A2BR signal mainly through Gs proteins resulting in the activation of adenylyl cyclase and an increase in intracellular cAMP levels. Of the adenosine subfamily, the A2BR subtype has been investigated least. Similar to all GPCRs, the adenosine A2BR is made up of seven transmembrane domains connected by three intracellular and three extracellular loops, an extracellular N terminus, and an intracellular C terminus. The A2BR has been implicated in several (auto)-immune diseases such as asthma and chronic obstructive pulmonary disease and is, therefore, an interesting drug target [1].
In a previous study, random mutagenesis combined with a yeast screen for activating mutant receptors have been performed on two parts of the adenosine A2BR in order to identify specific residues involved in the activation of the receptor [2, 3]. Mutations were randomly introduced in two separate fragments of the receptor, one ranging from the ATG until a KpnI restriction site in the second intracellular loop and a second form the BglII restriction site in the third intracellular loop until the end of the receptor (Fig. 1). These studies revealed many constitutively active mutations, both in the transmembrane domains as well as in the extracellular regions. The fragment that is in between the two restriction sites KpnI and BglII, encompassing transmembrane domain 4 (TM4), the second extracellular loop 2 (EL2), and transmembrane domain 5 (TM5), has so far not been examined. However, this region may be very important in the activation mechanism of the receptor. All three domains in this fragment have been implied to participate in the dynamic movements the receptor undergoes during activation [4]. Clear evidence for the importance of this region is also provided by the recently published crystal structures of both the antagonist- and agonist-bound adenosine A2AR, the closest homologue of the A2BR. In particular, EL2 and TM5 appear to be involved directly in ligand binding and in conformational movements induced by agonist binding [5–7]. Also, EL2 has been proposed to act as a negative regulator for the receptor, keeping it in its inactive state and is in many receptors part of the ligand binding site [8–11].

Snake plot of the adenosine A2BR. Mutated residues within the A2BR identified to result in increased constitutive activity are indicated in gray. These mutations originate from two previously described screens [2, 3] and the TM4-EL2-TM5 screen described in the current paper. The putative disulfide bridges are indicated with dotted lines. The disulfide bridge conserved in many class A GPCRs links C783.25 and C171EL2. The non-conserved second disulfide bridge between EL1 and EL2 is based on an analogous bond in the crystal structures of the adenosine A2AR (PDB: 3EML, 3QAK, 3YDO, and 3YDV); it links C72EL1 and C167EL2. The restriction sites KpnI and BglII are indicated; they were used to obtain the fragment for random mutagenesis
In the present study, we examined the influence of EL2 and its two adjacent transmembrane domains, TM4 and TM5, of the adenosine A2BR is made up of seven on receptor activation. We performed an unbiased random mutagenesis screen for gain-of-function mutations as well as constitutively active mutants (CAMs), i.e., mutant receptors that show basal activity independent of an agonist. In these CAMs, the equilibrium between the inactive (R) and active conformation (R*) is shifted, so that the active state is energetically more favorable than in the wild-type situation, similar to what occurs when the receptor binds an agonist [12]. Residues that are mutated to cause this shift in equilibrium are therefore likely to be involved in the on-and-off switch of the receptor and can provide us with information on the activation mechanism of the receptor.
We performed the screen for activating mutant receptors using a Saccharomyces cerevisiae strain that has been genetically modified to serve as a reporter system with growth as an output parameter. This yeast system is an ideal background to monitor activation of a single GPCR since the endogenous GPCR (Ste2) has been removed from the system while still maintaining the complete GPCR signaling machinery [13]. Several previous reports have proved this eukaryotic system to be predictive of the mammalian situation, as shown by functional and binding studies in CHO cells [2, 14]. Several mutated residues were identified, causing the receptor to be highly increased in both agonist-induced and spontaneous activity. The results presented here can be of general interest in increasing our understanding of the activation mechanism of class A GPCRs, of adenosine receptors in particular as well as other members of this superfamily.
Materials and methods
DNA constructs
The S. cerevisiae expression vector containing the adenosine A2BR gene, the pDT-PGK_A2BR plasmid, was kindly provided by Dr. Simon Dowell from GSK (Stevenage, UK). A KpnI restriction site was introduced in the A2BR gene in the region encoding the second intracellular loop. Together with a restriction site in the third intracellular loop, BglII, it is possible to divide the receptor into three large fragments suitable for random mutagenesis. The fragment between these two restriction sites was used in the mutagenic PCR. This fragment encodes transmembrane domain four, the second extracellular loop, and transmembrane domain five (TM4-EL2-TM5).
Mutagenic PCR for the construction of the random mutagenesis library
The introduction of random mutations in the adenosine A2BR was achieved by manipulating the polymerase chain reaction adapted from the method of Fromant et al. [15]. In this error-prone PCR method, the balance between Mg2+ ions and Mn2+ ions was shifted, compromising fidelity of the DNA polymerase enzyme. Furthermore, the introduction of mutations can be guided by adding excess of one of the nucleotides (the “forcing” nucleotide). This technique makes it possible to introduce mutations in fragments of up to 400 bp in length. The DNA fragment encoding TM4-EL2-TM5 encompasses 262 bp.
The mutagenic reaction contained 10 ng of template DNA, 0.1 μM concentrations of each primer, 0.2 mM concentrations of dNTPs as well as 3.4 mM concentrations of the nucleotide in excess dCTP, 0.5 mM MnCl2, 4.7 mM MgCl2, and 0.5 units of Super Taq polymerase without proofreading. The number of mutagenic PCR cycles was set to 10. Using these conditions, only a limited amount of mutations are introduced per fragment [16].
The following primers were used:
-
5′-GGTATAAAAGTTTGGTCACGGGTACCCGAGCAA-3′
-
5′-GAAGCTGCCTGCAGGCCACCAGGAAGATCTTAATG-3′
The mutagenic PCR products were submitted to agarose gel electrophoresis and the gel bands containing the mutated fragments were isolated from the gel and purified. Subsequently, the mutated fragments were amplified further with ten cycles of a regular PCR with the same primer sets. After the error-prone PCR, the normal fragment TM4-EL2-TM5 in the wild-type receptor was replaced by the mutated fragments using the restriction sites KpnI and BglII and transformed into DH5α Escherichia coli-competent cells (Invitrogen, San Diego, CA, USA). Plasmids were isolated from the culture resulting in a mutagenic adenosine A2BR library.
Transformation in MMY24 S. cerevisiae strain
pDT-PGK_A2BR plasmids were transformed into an S. cerevisiae yeast strain according to the Lithium-Acetate procedure [17]. The MMY24 strain is derived from the MMY11 strain and was further adapted to communicate with mammalian GPCRs through the introduction of a chimeric G protein [13]. The genotype of the MMY24 strain is: MATahis3 leu2 trp1 ura3can1 gpa1_::G_i3 far1 ::ura3 sst2_::ura3 Fus1::FUS1-HIS3 LEU2::FUS1-lacZ ste2_::G418R. To measure signaling of GPCRs, the pheromone signaling pathway of this strain was coupled via the FUS1 promoter to HIS3, a gene encoding the key enzyme in histidine production, imidazole glycerol-phosphate dehydrase. The degree of receptor activation was measured by the growth rate of the yeast on histidine-deficient medium. A second reporter gene was placed under control of the FUS1 promoter; the LacZ gene. Transcription of this gene results in the production of the enzyme β-galactosidase. The presence of this enzyme is also a measure of receptor activation.
Random mutagenesis screen
The random mutagenesis screen was performed on selection agar medium in two steps. Firstly, yeast cells were selected for the presence of the plasmid pDT-PGK using selection medium lacking the markers uracil and leucine. After 3 days of incubation at 30°C, positive colonies were pooled. For the second selection step, 104 cells were spread onto selection medium lacking uracil, leucine, and histidine (YNB-ULH) to select for the MMY24 yeast strain, the pDT-PGK plasmid, and activity of the receptor, respectively. A concentration of 7 mM 3-aminotriazole (3-AT), a competitive inhibitor of imidazole glycerol-phosphate dehydrase, was added to the agar plates to suppress basal yeast growth that occurs in histidine-deficient medium. Also, a concentration of 1 nM of the full A2BR agonist 5′-N-ethylcarboxamidoadenosine (NECA) was added to the medium, a concentration at which yeast cells expressing the wild-type human adenosine A2BR still barely grow. After 3 days, colonies were selected and transferred to new selection plates. To further select for true active mutant receptors, we performed a qualitative β-galactosidase assay according to protocol #PT3024-1 from Clontech (Clontech Laboratories, Mountain View, CA, USA). In brief, yeast colonies were transferred to a filter and lysed using repeated freeze-thawing with liquid nitrogen. The filter was then placed on top of several Whatman papers presoaked in Z-buffer (16.1 g/L Na2HPO4, 5.5 g/L NaH2PO4, 0.75 g/L KCl, 0.246 g/L MgSO4, and 0.3% β-mercaptoethanol) and 0.3 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactosidase), a substrate of β-galactosidase. The filter was incubated at 30°C until a blue color appeared. Colonies with a blue color were chosen for further characterization. Mutated receptors were sequenced and subsequently retransformed into the yeast strain to confirm their activated phenotype.
Liquid growth assay
To characterize the mutant receptors further, concentration-growth curves were generated in a liquid growth assay. This assay is on 96-wells scale and growth is easily determined by measuring absorption at a wavelength of 595 nm. In this assay, 150 μl liquid YNB-ULH medium with 7 mM 3-AT and a varying concentration of NECA (10−10–10−5 M) was added to each well. A concentration range from 10−10 to 10−5 M was also used for the concentration-growth curves with BAY 60-6583 (2-[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]pyridin-2-ylsulfanyl]acetamide; synthesized in house), a non-nucleoside agonist [18]. Yeast cells from an overnight culture were diluted to around four·106 cells/ml (OD600 ≈ 0.2) and 50 μl was added per well. The 96-well plate was then incubated for 35 h in a Genios plate reader (Tecan, Durham, NC, USA) at 30°C, keeping the cells in suspension by shaking every 10 min at 300 rpm for 1 min. Results originate from three independent experiments, performed in duplicate.
Solid growth assay
To monitor the response of the mutant receptors in the presence of the inverse agonist ZM241385 (4-{2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-yl-amino]ethyl}phenol), a solid yeast growth assay was used. Yeast growth was determined based on growth density, rather than absorption at 595 nm; this enabled us to visualize the level of constitutive activity more clearly. In the solid growth assay, yeast cells from an overnight culture were diluted to around 400,000 cells/ml (OD600 ≈ 0.02), and droplets of 1.5 μl were spotted on selection agar plates, YNB-ULH, containing 7 mM 3-AT and a ZM241385 concentration ranging from 10−11 to 10−5 M. For the single-point experiments, a final concentration of 10−5 M ZM241385 was used [19]. After incubation at 30°C for 50 h, the plates were scanned and receptor-mediated yeast growth was quantified with Quantity One imaging software from Bio-Rad (Hercules, CA, USA). The growth rate of yeast was calculated as the density of each spot with a correction for local background on the plate. Results are obtained from four independent experiments, performed in quadruplicate.
Whole cell radioligand binding experiments
Yeast cells expressing wild-type or mutated A2BRs were cultured overnight in rich YAPD (Yeast Extract Adenine Peptone Dextrose) medium. Cells were centrifuged for 5 min at 2,000 × g, the pelleted cells were once washed with 0.9% NaCl. The cells were again centrifuged 5 min at 2,000 × g and diluted in the assay buffer (50 mM Tris–HCl pH7.4 + 1 mM EDTA) to OD600 = 40 (OD600 = 1 ≈ 2.5·107 cells/ml). Binding experiments were performed with 1.3 nM [3H]PSB-603 (8-[4-[4-(4-chlorophenyl)piperazine-1-sulfonyl)phenyl]]-1-propylxanthine; K D, yeast = 0.8 ± 0.02 nM) and a final cell concentration of 25·107 cells/ml in a total volume of 100 μl [3, 20]. Nonspecific binding was determined in the presence of 1 mM NECA. For whole competition curves, a concentration range of 10−10–10−3 M of the ribose agonist NECA, 10−10–10−4 M of the non-ribose agonist BAY 60-6583, or 10−10–10−4 M of the inverse agonist ZM241385 was used. Samples were incubated for 1 h at 25°C while shaking vigorously to keep the yeast cells in suspension. Incubation was terminated by adding 1-ml ice-cold assay buffer. Bound from free radioligand was immediately separated by rapid filtration through Whatman GF/B filters pre-incubated with 0.1% polyethylenimine using a Millipore manifold during which the filters were washed six times with ice-cold assay buffer. Filter-bound radioactivity was determined by scintillation spectrometry (Tri-Carb 2900TR; PerkinElmer Life and Analytical Sciences) after addition of 3.5 ml of PerkinElmer Emulsifier Safe. Results are obtained from three independent experiments, performed in duplicate.
Whole cell extracts and immunoblotting
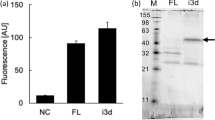
Whole protein cell extracts were made from the transformed yeast cells using trichloroacetic acid (TCA). From an overnight culture, 1.2·108 yeast cells were harvested in mid-log phase. The cells were washed twice with 20% TCA after which they were broken by vigorous vortexing in the presence of glass beads. The yeast cell extracts were separated using SDS/PAGE and subsequently blotted on Hybond-ECL membranes. For this purpose, a sample of 4.0 μl containing 12 μg protein was loaded on a 12.5% SDS/PAGE gel. A semi-automated electrophoresis technique (PhastSystem™, Amersham Pharmacia Biotech) was used for SDS/PAGE as well as blotting. The antibody directed against the C-terminal region of the adenosine A2BR was kindly provided by Dr. I. Feoktistov (Vanderbilt University, Nashville). Densitometric analysis of the protein bands was performed using the volume analysis tool as present in the Quantity One imaging software from Bio-Rad (Hercules, CA, USA). The nonspecific band at approximately 45 kDa was used as loading control. The ratio between specific A2BR protein bands (at 29 and 48 kDa) and the nonspecific band was determined and the wild-type receptor was set at 100%, the empty vector pDT-PGK at 0%. The experiment was performed in duplicate. The relative expression levels represent expression of the whole receptor population, not distinguishing between cell surface and intracellularly expressed protein
Bioinformatics—mapping the mutated residues onto the adenosine A2AR structure
The positions of the mutations identified in the random mutagenesis screen were mapped onto the corresponding positions in the crystal structure of the agonist-bound adenosine A2AR (PDB: 2YDV) [7]. These corresponding positions were determined by a multiple sequence alignment created using ClustalW with default parameters. To make use of known crystallographic data, the sequences of the CXCR4 chemokine receptor (CXCR4R) and the β2-adrenergic (b2AR) receptor were included in this alignment. The EL2 was defined from the crystal structures of the A2AR (residues, 143–173), the CXCR4R (residues, 175–192), and the b2AR (residues, 171–196). The amino acids in the TM domains of the various receptors were related through their Ballesteros and Weinstein numbering [21].
Bioinformatics—cysteine occurrence analysis
To gain insight in the number of class A GPCRs that have multiple cysteines present in the second extracellular loop, we analyzed all human olfactory (415) and non-olfactory (221) class A GPCRs as present in the GPCRDB [22]. A cysteine count was performed on the second extracellular loop that for this analysis was defined as the fragment between residues 4.55 and 5.38 according to the Ballesteros and Weinstein numbering [21]. These two residues are located at the interface of the loop and the transmembrane domains but are present within the membrane in all predictions.
Results
Random mutagenesis screen in yeast
Mutations were randomly introduced in the fragment encoding for TM4, the EL2, and TM5 of the human adenosine A2BR using an error-prone PCR reaction (Figs. 1 and 2). The corresponding fragment in the wild-type receptor was replaced after mutagenesis by the mutated fragments, which rendered a set of approximately 5,000 different plasmids of which ca. 80% contained mutations. The mutations occurred in a low frequency; most of the mutant receptors contained single-point mutations and the mutant receptor with the highest mutation frequency contained five nucleotide changes. These results are comparable to a random mutagenic library of the first three transmembrane domains of the A2BR published by Beukers et al. [2]. After ligation, the plasmids were propagated in competent E. coli cells, resulting in the final mutant A2BR library (Fig. 2). This mutant library was subsequently transformed in the S. cerevisiae MMY24 strain. The MMY24 yeast strain has been genetically modified to enable mammalian GPCRs to couple to the yeast pheromone pathway with subsequent transcription of the reporter gene HIS3, increasing the histidine synthesis. We used this trait to specifically select yeast cells that express an active receptor by their ability to produce histidine and therefore grow on histidine-deficient medium. Before starting the actual screen, we first selected yeast cells that were successfully transformed. We then screened the transformed MMY24 cells for an active phenotype on selection plates lacking YNB-ULH to select for the MMY24 yeast strain, the pDT-PGK plasmid, and activity of the receptor, respectively. By also adding a low concentration of 1 nM of the agonist NECA (EC50 value at wild-type A2BR: 137 ± 10 nM, see also Table 1), we screened for both CAMs and mutant receptors with increased agonist potency. In total, ca. 0.5 million yeast clones were used for the final activation screen, so the library was screened approximately 100 times. From the screen, all well separated colonies that appeared on the selection plates were selected (a total of 148 yeast colonies).

Schematic overview of the screen for activating mutations. The mutated fragments from the mutagenic PCR were reintroduced in an otherwise wild-type A2BR in the pDT-PGK vector, resulting in the mutagenic A2BR library. The library was transformed in the MMY24 yeast strain and screened for mutant receptors with constitutive activity and/or an increased potency for NECA. As a second selection criterion, the presence of β-galactosidase was determined in a qualitative assay, resulting in a final selection of 35 yeast colonies. Sequencing revealed 12 different mutant receptors
Besides the reporter gene HIS, a second reporter gene was incorporated into the MMY24 genome under the same promoter; the LacZ reporter gene, causing the yeast cell to also produce the enzyme β-galactosidase. To proceed with the most active mutant receptors, we performed a qualitative β-galactosidase assay. The colonies were lysed and the presence of the enzyme β-galactosidase was measured. The 35 colonies with the strongest response in this assay, out of the original 148, were selected, plasmids were isolated from the yeast cells, and the mutations were identified by sequencing. Several of the nucleotide changes observed in the sequences resulted in the same amino acid changes in the A2BR. Mutant receptor G135A4.55/I197L5.53/Y202N5.58 was identified most, namely 12 times out of the 35 sequenced plasmids (shown in superscript are the positions according to the Ballesteros and Weinstein GPCR numbering system [21]) (Fig. 2; Table 1).
In total, 12 different mutant receptors were identified, containing altogether 13 mutated positions (Fig. 1). Among the 12 mutant receptors, residues F1414.61, C167EL2, and Y2025.58 were found to be mutated more than once. Amino acid changes of F1414.61 were identified in two different receptors; in the single mutant F141L4.61 and in the double mutant F141C4.61/Y202C5.58. The mutation C167SEL2 was found as a single mutant as well in combination with a residue outside of the cluster: T155EL2. Mutations at position Y2025.58 were present in three different mutant receptors: G135A4.55/I197L5.53/Y202N5.58, F141C4,61/Y202C5.58, and Y202S5.58 (Table 1).
The mutated residues form three distinct clusters in the receptor; at the top of TM4, at a cysteine-rich area in EL2, and at the bottom of TM5. Four receptors contained mutations in TM4, forming a small cluster of three amino acids: G1354.55, I364.56, and F1414.61. In EL2, a total of seven residues were found mutated in five different mutant receptors. Five of the seven residues form a tight cluster, namely C166EL2, C167EL2, L168EL2, V169EL2, and F173EL2. The cluster seen in TM5 consists of three residues: I1975.53, V2005.56, and Y2025.58 (Fig. 1; Table 1).
Constitutively active and gain-of-function mutants
To confirm the active phenotype, all 12 receptors were retransformed into the yeast strain and their pharmacology was investigated using yeast growth assays. All mutant receptors showed an increase in constitutive activity, although less pronounced in mutant receptors F141L4.61 and I136T4.45 (1.5 times compared with wild type) (Fig. 3a; Table 1). Mutant receptor G135A4.55/I197L5.53/Y202N5.58 showed the largest increase in basal activity with yeast growth levels 38-fold over wild type, corresponding to 62% of the maximal activation level. The constitutive activity of this mutant receptor could be reduced by the inverse agonist ZM241385, however, a residual activity of 8% remained. The level of constitutive activity of the other 11 mutant receptors could be fully suppressed (Fig. 3a). For mutant Y202S5.58, a full concentration-growth inhibition curve with ZM241385 showed a potency of 31.3 ± 4.4 nM for ZM241385. In comparison, in literature values between 13 and 50 nM have been reported for wild-type A2BR [23, 24] (Fig. 3b).

Constitutive activity of the mutant receptors determined in a solid yeast growth assay. a Growth was measured on selective agar plates YNB-ULH in the absence of a ligand or in the presence of 10 μM of the inverse agonist ZM241385. The vertical bars in the graph represent the percentage spontaneous activity of wild-type A2BR and mutant receptors (black) and the response to ZM241385 (white). The maximal activation level of the wild-type receptor was set at 100%; the background of the selection plate was set at 0%. b Concentration-growth inhibition curve of mutant receptor Y202S5.58. A concentration range of 10−11–10−5 M ZM241385 was added to the selection agar plates. The figure is representative of one experiment performed in quadruplicate. The IC50 value for ZM241385 was 31.3 ± 4.4 nM, determined from four independent experiments
Concentration-growth curves revealed that all mutant receptors showed a large increase in potency of more than one log-unit for the full agonist NECA (Table 1). Curves of mutant receptors F141L4.61, C167SEL2, Y202S5.58, G135A4.55/I197L5.53/Y202N5.58, and T155AEL2/C167SEL2 are shown in Fig. 4. Even though the level of constitutive activity in mutant G135A4.55/I197L5.53/Y202N5.58 was very high, the maximal level of receptor activation could be reached in response to NECA in a dose-dependent manner, with an EC50 value of 7.1 ± 0.7 nM compared with 137 ± 10 nM seen for wild-type A2BR (Table 1). Mutant receptor Y202S5.58 also has a relatively high level of constitutive activity being 13-fold higher than wild-type receptor. However, upon stimulation with NECA the maximum receptor activation (E max) was lower than observed with wild-type A2BR. Similar results were observed for the receptor where Y2025.58 was mutated in combination with residue F141: mutant receptor F141C4.61/Y202C5.58 (Table 1).

Concentration-response curves from liquid yeast growth experiments. Curves are shown of wild-type (filled squares) and mutant receptors F141L4.61 (filled triangles), C167SEL2(inverted filled triangles), Y202S5.58 (filled circles), G135A4.55/I197L5.53/Y202N5.58(filled diamonds), and T155AEL2/C167SEL2 (empty circles). Curves are shown from a representative experiment. Data for all experiments are presented in Table 1
Residues F1414.61 (TM4), C167EL2 (EL2), and Y2025.58 (TM5) were identified multiple times in the screen, suggesting a particular important function of these positions in receptor activation. For additional studies, we therefore focused on the single mutant receptors containing each of the residues; F141L4.61, C167SEL2, and Y202S5.58. These single mutants were also studied with BAY 60-6583, a structurally different A2BR agonist [18]. This full agonist lacks a ribose moiety that is present in the adenosine derivative NECA and previously thought to be essential for adenosine receptor activation. The chemical structures of both agonists NECA and BAY 60-6583 are shown in Tables 1 and 2. BAY 60-6583 was more than five times more potent at mutant receptor F141L4.61; in comparison, NECA’s potency was more than 25-fold increased. Both NECA and BAY 60-6583 displayed a 10-fold increase in potency on mutant Y202S5.58. Mutant receptor C167SEL2 showed a small 2-fold increase in potency in response to BAY 60-6583, with NECA this was over 15-fold.
Western blot analysis of whole cell lysates showed a similar degree of expression of the mutant receptors compared with the wild-type receptor, indicating that the increase in activation profile was not due to overall increases in receptor levels in the system, both intracellular and expressed at the cell surface. Results of mutant receptors F141L4.61, C167SEL2, and Y202S5.58 are shown in Fig. 5.

Western blot analysis. The relative expression of mutants F141L4.61, C167SEL2, and Y202S5.58 compared with the wild-type receptor was determined using densitometric analysis of the Western blots. The ratio was determined between the density of the specific bands and the density of the nonspecific band that is always present on the blot. Wild-type receptor was set at 100%; the empty vector pDT-PGK was set at 0%. The experiment was performed in duplicate. One representative blot is shown in the upper panel. The arrows indicate the specific A2BR bands; these are located at ca. 29 and 48 kDa
Radioligand binding experiments on the activated mutant receptors
To investigate the influence of the mutated residues on binding next to activation, we performed radioligand binding experiments using the A2BR selective antagonist [3H]PSB-603 [3, 20]. Saturation binding experiments showed a K D of the radioligand on whole yeast cells expressing the hA2BR of 0.81 ± 0.02 nM (data not shown), in comparison, the K D value determined on CHO cell membranes expressing the hA2BR was 0.40 ± 0.19 nM [20]. Competition binding curves were made for wild-type and mutant receptors with unlabeled NECA, BAY 60-6583, or ZM241385 as a displacer. For mutant receptor Y202S5.58, we were not able to reach a high enough window to perform competition binding curves. This is likely due to a decrease in affinity for the radiolabeled antagonist [3H]PSB-603 since no change in expression levels was observed (Fig. 5). Mutant receptors F141L4.61 and C167SEL2 showed a large increase in NECA affinity compared with the wild-type receptor of 13- and 9-fold, respectively (Table 2). In the radioligand binding experiment with BAY 60-6583 as displacer, the affinity of mutant F141L4.61 was again increased. In contrast, mutant C167SEL2 had an affinity for BAY 60-6583 similar to wild-type A2BR (Fig. 6; Table 2). The affinity of the inverse agonist ZM241385 was also determined. Both mutant receptors did not show a large change in affinity for ZM241385 compared with the wild-type receptor, although the effect on F141L4.61 was significant (Table 2).

Competition binding curves using [3H]PSB-603. Displacement curves of wild type A2BR (filled squares), mutant receptor F141L4.61 (inverted filled triangles) and mutant receptor C167SEL2 (filled triangles) with the ribose agonist NECA (a) and the non-ribose agonist BAY 60-6583 (b). The graphs are the combined result of three independent experiments performed in duplicate
Bioinformatic analyses
Recently, several crystal structures of the adenosine A2AR have been elucidated (PDB entry codes: 3EML, 3QAK, 2YDO, 2YDV) [5–7]. The adenosine A2AR is the closest homologue of the adenosine A2BR, with 82.1% amino acid similarity and 59.4% identity. We mapped the mutated residues onto the crystal structure of the active A2AR structure bound to NECA (PDB: 2YDV) in order to obtain a view on the 3D localization of the residues (Fig. 7). The corresponding positions of the mutated A2BR residues in the A2AR sequence were determined by a multiple sequence alignment (Fig. 7a). To improve the quality of the multiple sequence alignment and to determine the transition between loop and transmembrane domains, we also included sequences of two other crystallized GPCRs; the b2AR receptor and the CXCR4R receptor [25, 26]. Also in the 3D view, clusters of mutations can be observed in the top of TM4 and the bottom of TM5 (Fig. 7a). The corresponding positions of the cluster in EL2 appear to reside in a part of the loop that is involved in forming an anti-parallel β-sheet with EL1. It is very likely that this typical protein structure is also present in the A2BR and constitutively active mutations in the putative beta-strand in EL1 of this receptor have previously been described by our laboratory [3].

Mutated residues of the A2BR mapped on the crystal structure of the A2AR. a Multiple sequence alignment with all four adenosine receptor subtypes (A2AR, A2BR, A1R, and A3R) as well as the sequences of two other crystal structures of CXCR4 and the b2AR to ensure the most plausible definition of EL2. The mutated residues are indicated in bold font and underlined. b Based on the multiple sequence alignment, the mutated residues identified in the random mutagenesis screen were mapped onto the crystal structure of the A2AR (PDB: 2YDV; blue), the closest homologue of the A2BR. The corresponding mutated residues and their side-chains are indicated in magenta. The ribbon representation also shows the co-crystallized ligand NECA as well as the three disulfide bridges that restrict EL2 in the A2AR
In the A2AR structure, all three available cysteines in the second extracellular loop are involved in bridge formation. It has been proposed that formation of extracellular disulfide bridges may be an important general mechanism for regulating the activity of GPCRs [27]. The adenosine A2BR has a high number of cysteines in EL2 that could all potentially form extracellular disulfide bridges. A previous sequence alignment analysis performed by de Graaf et al. already revealed that over 90% of class A GPCRs contain at least one cysteine in EL2 and that several receptors contain more. In most cases, the most downstream cysteine appeared involved in forming the conserved disulfide bridge with TM3 [28]. To investigate how common multiple cysteines are in class A GPCRs, we performed a cysteine occurrence analysis. We counted the number of cysteines present between residues 4.55 and 5.38 [21], a region that encompasses the second extracellular loop. The majority of non-olfactory receptors contains only one cysteine within EL2 (Fig. 8a); this cysteine represents in most cases the conserved cysteine present in over 90% of class A GPCRs. From what is known from the currently available crystal structures this conserved cysteine forms a disulfide bridge with a cysteine present in the top of TM3 that is essential for receptor structure and function. The adenosine A2BR is the only receptor with four cysteines in EL2, which is the highest cysteine count in this analysis. The adenosine A2AR, the closest homologue of the A2BR, contains one cysteine less with three cysteines in the loop. The other adenosine receptor subtypes, the A1R and A3R, only hold one cysteine in EL2. In EL2 of olfactory receptors, generally multiple cysteines are present; ca. 80% of the receptors contain three cysteines in the loop (Fig. 8b). This special subfamily of class A GPCRs is responsible for our sense of smell by binding odorants. Metal ions, such as zinc, have been proposed to be essential in recognition and binding of odorants to their receptor, in which ligation of the metal ion to the thiol group of cysteine residues might play an important role [29].

Cysteine occurrence analysis. The number of cysteines present in the second extracellular loop was counted in human class A GPCRS. For this analysis, the loop was defined between residues 4.55 (TM4) and 5.38 (TM5). The graphs represent all human class A GPCRs, either non-olfactory (a) or olfactory (b), as present in the GPCRDB (http://www.gpcr.org/7tm/), with the x-axis the number of cysteines counted in EL2. The majority of non-olfactory receptors contains one cysteine in EL2; the adenosine A2BR alone is responsible for the bar at four cysteines. Most olfactory receptors have three cysteines in the loop
Discussion
A random mutagenesis screen for gain-of-function and CAMs was performed on fragment TM4-EL2-TM5 of the human adenosine A2BR. These three regions of the receptor have been implied to participate in the dynamic movements the receptor undergoes during activation. Upon receptor activation, a coupling between movements of EL2 and TM5 has been observed as well as a rearrangement in the hydrogen-bonding networks connecting EL2 with the extracellular ends of TM4, TM5 and TM6 [4].
For the β1-adrenergic receptor, the β2-adrenergic receptor, (rhod)opsin, and the adenosine A2AR, we now have access to crystal structures of both inactive and active conformations [5–7, 30–35]. These structures reveal that in the transition between the inactive and the active conformation subtle changes at the extracellular surface and the ligand binding site lead to large movements at the intracellular surface. The lower regions of TM5 and TM6 show a particularly large displacement that is allowed by the presence of conserved prolines (5.50 and 6.50) that interrupt the hydrogen bond network within the helices.
From the screen, 12 different mutant receptors were identified. Most of these mutants show a significant increase in constitutive activity, with mutant G135A4.55/I197L5.53/Y202N5.58 even reaching a basal activity that is over 60% of the maximal activation level (Fig. 3). All mutant receptors displayed a very large increase in potency for NECA compared with wild type, ranging from an improvement in activation of 11-fold (V200G5.56) to even 25-fold (F141L4.61) (Table 1). That we were able to identify mutant receptors with such large effects on activation further emphasizes the strength of using an unbiased random mutagenesis approach in combination with the S. cerevisiae system.
Three “hotspots” important for A2BR activation
The residues found mutated in our screen are located in three distinct clusters: at the top of TM4, in a cysteine-rich region in EL2, and at the bottom half of TM5 (Figs. 1 and 7b). Even though a number of mutant receptors contain multiple amino acid changes, no combinations between mutations in EL2 and the transmembrane domains were identified. This suggests that the influence of EL2 on receptor activation is at a different level than that of the transmembrane domains. There is accumulating evidence that agonist binding and activation occur through a series of conformational intermediates, for which multiple switches are needed to be activated [36, 37]. It is quite likely that the first switch is present at the site of ligand entry to the receptor, i.e., at the extracellular surface.
The cluster in TM4 consists of three amino acids; G1354.55, I1364.56, and F1414.61. A saturated mutagenesis screen performed on the transmembrane domains of the complement factor 5a receptor, also revealed important residues at the extracellular membrane interface of TM4, corresponding to positions 4.46, 4.53, 4.55, 4.57, 4.58, and 4.60 [38, 39]. Very recently, Warne et al. published four structures of the β1-adrenergic receptor bound to full and partial agonists. One difference between an antagonist and a full agonist-bound β1-adrenergic receptor is that a van der Waals interaction is broken between positions 4.56 and 5.46. This results in a reduced interface between helix 4 and 5 that may be significant in the activation process [30]. In the inactive structure of the adenosine A2AR (PDB/3EML), a similar van der Waals interaction exists between I1354.56 and C1855.46. Also, a hydrogen bond is formed between Q893.37 and the backbone of C1855.46 [5]. In the active structures of the A2AR (PDB: 3YDO/3YDV/3QAK), the cysteine side chain shifts and the hydrogen bond is broken [6, 7]. Noteworthy is that in the 3YDO and 3YDV structures bound to adenosine and NECA, respectively, position 3.37 was mutated (Q89A) [7].
The mutations found mutated in EL2 are located in a cysteine-rich region of the loop that may be involved in a β-strand structure in the loop as is seen in the structure of the A2AR (Fig. 7b). The partnering strand in EL1 that is involved in forming the β-sheet with EL2 has previously been reported to be essential in A2BR activation [3]. EL2 has been suggested to act as a negative regulator that keeps the receptor in a silent state before agonist-induced activation [8–10]. The mutant receptors in EL2 that we identified all showed a large increase in receptor activation, both in response to the agonist NECA and independent of a ligand. This could suggest that the cysteine-rich cluster we identified in our screen has a similar regulating role in suppressing receptor activation in its basal state. The cysteine occurrence analysis on all human class A GPCRs revealed that the A2BR contains an exceptionally large number of four cysteines in EL2, whereas most non-olfactory class A GPCRs only contain one cysteine (Fig. 8; see also an analysis by de Graaf et al. [28]). In olfactory receptors, the cysteine occurrence is much higher, which may be linked to metal ion binding [29].
In TM5, the mutated residues are found in a small cluster of three residues at the bottom of the helix: I1975.53, V2005.56, and Y2025.58. From the crystal structure of the A2AR, as well as additional mutagenesis data, several residues in TM5 have been indicated to play a role in agonist and/or antagonist binding: M1775.38, F1805.41, N1815.42, and F1825.43 [5, 40]. The recently published active structures in which NECA and adenosine are co-crystallized, reveal that M1775.38, N1815.42 are directly involved in the ligand binding pocket of these ribose-containing agonists [7]. The residues identified in our screen are located much lower in the helix and are therefore unlikely to participate in the ligand binding pocket. However, the large increase in agonist potency and constitutive activity observed in our study implies that these residues are essential in receptor activation.
CAMs in the adenosine A2BR
The fragments upstream and downstream of the investigated fragment TM4-EL2-TM5 had been subjected to a random mutagenesis screen in a similar manner in our laboratory [2, 3]. In Fig. 1, the ensemble of CAM residues identified in the different studies is indicated in gray, from which we conclude that next to the described clusters in TM4, EL2, and TM5, similar series of residues exist in TM3 and TM6. Even though the screening set up in all three screens (ATG-KpnI, TM4-EL2-TM5, and BglII-stop) was chosen such that both CAMs and gain-of-function mutants would be selected, all the identified mutant receptors displayed an increase in constitutive activity. The levels of constitutive activity ranged from a 1.5-fold change (F141L4.61 and N36DIL1) compared with the wild-type receptor to an immense increase of 38-fold for mutant receptor G135A4.55/I197L5.53/Y202N5.58. Interestingly, of all 41 residues identified in constitutively active mutants of the A2BR, only four were actually involved in binding of either adenosine or NECA at the corresponding positions in the crystal structures of the A2AR published by Lebon et al. [7] These corresponding residues in the A2BR are: F173EL2, V2506.51, N2546.55, and T2576.58. The last three, all residing in TM6, were identified in one particular multiple mutant receptor; Q214L/I230N/V240M/V250M/N254Y/T257S/K269stop (IL3/6.31/6.41/6.51/6.55/6.58/EL3) [2]. The residue F173EL2 (F168 in A2AR) directly interacts with all the ligands co-crystallized with the A2AR through a π-stacking contact. For the antagonist ZM241385, the triazolotriazine ring interacts with the phenylalanine, where in the agonists UK-432097, NECA, and adenosine, π-stacking occurs with the adenine moiety. In the study described here, the F173LEL2 mutant receptor displayed a large increase in potency for NECA. Mutating the corresponding F168EL2 in the A2AR into other aromatic residues resulted in a moderate decrease in activation of the receptor; mutation into an alanine virtually abolished activation [40]. Assuming that the F173EL2 in the A2BR has a similar role in binding, a large hydrophobic residue might be required at this position, however removing the aromatic side chain can even improve agonist access to the binding pocket.
Many of the CAMs in our studies only have one or two amino acid changes, indicating that quite subtle changes can lead to a large impact on the receptor activation mechanism and that these residues are not necessarily directly involved in either ligand binding or G protein coupling.
Residues F1414.61, C167EL2, and Y2025.58
The amino acids F1414.61, C167EL2, an Y2025.58 were identified multiple times in the screen. This may indicate that these residues are of particular importance in the activation mechanism of the A2BR. Position F1414.61 has been reported previously as being involved in affinity and potency changes. The polymorphic variant M172I4.61, located at the corresponding position in the serotonin 1A receptor, displayed a 3-fold increase in agonist potency [41]. In another subtype of serotonin receptors, 5HT1B, substitution of the amino acid F1854.61 by an alanine increased the affinities for several agonists [42]. In the study described here, mutating F1414.61 to a leucine resulted in a 25-fold increase in potency for NECA and a 5-fold increase in potency in response to the non-ribose agonist BAY 60-6583 (Fig. 2, Table 1). Radioligand binding studies revealed that affinity for both agonists was also largely increased (Table 2). The residue is located at the onset of EL2, pointing outwards (Fig. 7b). Although the residue is at great distance from the putative binding pocket of both NECA and BAY 60-6583, it is firmly involved in both agonist activation and binding [43]. The location of the residue does suggest a main role in positioning EL2, and could therefore be indirectly involved in shaping the entry of the agonist binding pocket.
Mutant receptor C167SEL2 showed an increase of ca. 16-fold in potency for the adenosine derivative NECA as well as a constitutive activity that was 6-fold higher compared with wild type (Table 1). Radioligand binding experiments also revealed an increase in binding affinity for the adenosine derivative NECA (Fig. 5). Interestingly, when we activated the C167SEL2 mutant with BAY 60-6583, a structurally different agonist that lacks a ribose moiety, only a 2-fold change in potency was observed and affinity remained unchanged compared with wild type (Fig. 5; Table 1). Residue C167EL2 is likely able to form a non-conserved disulfide bridge with a cysteine in EL1 in the A2BR as seen in the crystal structure of the closest family member, the A2AR [3] (Fig. 1). Our results indicate that the putative disulfide bridge between C167EL2 and C72EL1 is important for ribose agonist binding and activation, but less so for non-ribose agonists. Schiedel et al. recently performed a site-directed mutagenesis study on the cysteine residues present in EL2 of the A2BR. Mutating C167EL2 to a serine resulted in a 2.5-fold increase in potency for BAY 60-6583, similar to our observations, although the response to NECA of this mutant receptor was less pronounced in their study [44]. The tyrosine at position 2025.58 is highly conserved among class A GPCRs (88%) and there are several studies reporting this position as being important in receptor activation and G protein signaling. A somatic mutation in the thyroid-stimulating hormone receptor involved in toxic adenoma, Y601N5.58, showed increased levels of constitutive activity, but was unable to couple to Gq/11 [45]. Very recently, Sansuk et al. proposed that movement at the extracellular side of TM5 is transduced as a set of structural rearrangements toward the intracellular side, so enabling interactions of Y5.58 with R3.50 in the cytoplasmic side of the receptor [46]. When comparing the inactive and active structures of the adenosine A2AR, we learned that Y5.58 (Y197 in the A2AR) displays a large rotameric shift upon activation. While in the ZM241385-bound, inactive structure the conserved Y1975.58 is located in between TM3 and TM6, this residue moves outward in the agonist-bound forms allowing TM5 to shift toward TM6. As a result, the intracellular ends of TM5 and TM6 move closer together in the active structures compared with the inactive structure, enabling access of the G protein [5–7]. In our screen, mutant Y202S5.58 showed a 13-fold increase in constitutive activity that could be reduced to wild-type levels in response to the inverse agonist ZM241385 with an IC50 comparable to the wild-type receptor, indicating that the mutation does not lock the receptor in an active conformation [23, 24] (Fig. 3). NECA potency was 11-fold increased, but maximal activation levels could not be reached, suggesting a decrease in coupling to and signaling through the G protein (Fig. 2; Table 1).
Concluding remarks
By applying an unbiased random mutagenesis approach with subsequent phenotype screening in a robust yeast system, we identified three hotspots in the A2BR that show a vast increase in both spontaneous and agonist-induced activity. None of the identified residues within these three clusters are part of the ligand binding pocket, yet, they are involved in agonist potency and affinity. Some of the identified residues, like C167EL2, most likely contribute to an A2BR-specific response to agonists. Others, such as F1414.61 and Y2025.58, might be part of a general activation mechanism for class A GPCRs. An overview of all the CAMs in the A2BR identified so far, indicates that there are several clusters of amino acids responsible for maintaining the subtle equilibrium that exist between the active conformation R * and the inactive conformation R of the receptor and that these residues are not necessarily directly involved in either ligand binding or G protein coupling. In more general terms, the results presented here could be of great use in unraveling the molecular details of GPCR activation.
References
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev 63(1):1–34. doi:10.1124/pr.110.003285
Beukers M, van Oppenraaij J, van der Hoorn P, Blad C, den Dulk H, Brouwer J, IJzerman A (2004) Random mutagenesis of the human adenosine A2B receptor followed by growth selection in yeast. Identification of constitutively active and gain of function mutations. Mol Pharmacol 65(3):702–710
Peeters MC, van Westen GJ, Guo D, Wisse LE, Muller CE, Beukers MW, IJzerman AP (2011) GPCR structure and activation: an essential role for the first extracellular loop in activating the adenosine A2B receptor. FASEB J 25(2):632–643. doi:10.1096/fj.10-164319
Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ, Smith SO, Eilers M (2009) Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol 16(2):168–175
Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, IJzerman AP, Stevens RC (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322(5905):1211–1217
Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science. doi:10.1126/science.1202793
Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474(7352):521–525. doi:10.1038/nature10136
Klco J, Wiegand C, Narzinski K, Baranski T (2005) Essential role for the second extracellular loop in C5a receptor activation. Nat Struct Mol Biol 12(4):320–326
Massotte D, Kieffer BL (2005) The second extracellular loop: a damper for G protein-coupled receptors? Nat Struct Mol Biol 12(4):287–288
Peeters MC, van Westen GJ, Li Q, IJzerman AP (2011) Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci 32(1):35–42. doi:10.1016/j.tips.2010.10.001
Jaakola VP, IJzerman AP (2010) The crystallographic structure of the human adenosine A(2A) receptor in a high-affinity antagonist-bound state: implications for GPCR drug screening and design. Curr Opin Struct Biol 20:1–14
Smit M, Vischer H, Bakker R, Jongejan A, Timmerman H, Pardo L, Leurs R (2007) Pharmacogenomic and structural analysis of constitutive G protein-coupled receptor activity. Annu Rev Pharmacol Toxicol 47:53–87
Brown A, Dyos S, Whiteway M, White J, Watson M, Marzioch M, Clare J, Cousens D, Paddon C, Plumpton C, Romanos M, Dowell S (2000) Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein alpha-subunit chimeras. Yeast 16(1):11–22
Stewart GD, Valant C, Dowell SJ, Mijaljica D, Devenish RJ, Scammells PJ, Sexton PM, Christopoulos A (2009) Determination of adenosine A1 receptor agonist and antagonist pharmacology using Saccharomyces cerevisiae: implications for ligand screening and functional selectivity. J Pharmacol Exp Ther 331(1):277–286
Fromant M, Blanquet S, Plateau P (1995) Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal Biochem 224(1):347–353
Beukers M, IJzerman A (2005) Techniques: how to boost GPCR mutagenesis studies using yeast. Trends Pharmacol Sci 26(10):533–539
Gietz D, St Jean A, Woods RA, Schiestl RH (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20(6):1425
Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK (2007) Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115(12):1581–1590
Li Q, Ye K, Blad CC, den Dulk H, Brouwer J, IJzerman AP, Beukers MW (2007) ZM241385, DPCPX, MRS1706 are inverse agonists with different relative intrinsic efficacies on constitutively active mutants of the human adenosine A2B receptor. J Pharmacol Exp Ther 320(2):637–645
Borrmann T, Hinz S, Bertarelli DC, Li W, Florin NC, Scheiff AB, Muller CE (2009) 1-alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J Med Chem 52(13):3994–4006
Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three dimensional models and computational probing of structure–function relations in G-protein coupled receptors. Meth Neurosci 25:366–428
Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, Klomp J, Oliveira L, de Vlieg J, Vriend G (2011) GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res 39(Database issue):D309–D319. doi:10.1093/nar/gkq1009
de Zwart M, Vollinga RC, Beukers MW, Sleegers DF, Kunzel JKVD, de Groote M, IJzerman AP (1999) Potent antagonists for the human adenosine A(2B) receptor. Derivatives of the triazolotriazine adenosine receptor antagonist ZM241385 with high affinity. Drug Dev Res 48(3):95–103
Ongini E, Dionisotti S, Gessi S, Irenius E, Fredholm BB (1999) Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 359(1):7–10
Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330(6007):1066–1071
Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318(5854):1266–1273
Storjohann L, Holst B, Schwartz TW (2008) A second disulfide bridge from the N-terminal domain to extracellular loop 2 dampens receptor activity in GPR39. Biochemistry 47(35):9198–9207
de Graaf C, Foata N, Engkvist O, Rognan D (2008) Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins 71(2):599–620
Wang J, Luthey-Schulten ZA, Suslick KS (2003) Is the olfactory receptor a metalloprotein? Proc Natl Acad Sci USA 100(6):3035–3039. doi:10.1073/pnas.262792899
Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG (2011) The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469(7329):241–244. doi:10.1038/nature09746
Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454(7201):183–187
Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469(7329):175–180. doi:10.1038/nature09648
Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK (2007) Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature 450(7168):383–387
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289(5480):739–745
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454(7203):486–491
Kobilka BK, Deupi X (2007) Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 28(8):397–406
Ahuja S, Smith SO (2009) Multiple switches in G protein-coupled receptor activation. Trends Pharmacol Sci 30(9):494–502
Baranski T, Herzmark P, Lichtarge O, Gerber B, Trueheart J, Meng E, Iiri T, Sheikh S, Bourne H (1999) C5a receptor activation. Genetic identification of critical residues in four transmembrane helices. J Biol Chem 274(22):15757–15765
Geva A, Lassere TB, Lichtarge O, Pollitt SK, Baranski TJ (2000) Genetic mapping of the human C5a receptor. Identification of transmembrane amino acids critical for receptor function. J Biol Chem 275(45):35393–35401. doi:10.1074/jbc.M005602200
Jaakola VP, Lane JR, Lin JY, Katritch V, IJzerman AP, Stevens RC (2010) Ligand binding and subtype selectivity of the human A(2A) adenosine receptor: identification and characterization of essential amino acid residues. J Biol Chem 285(17):13032–13044. doi:10.1074/jbc.M109.096974
Del Tredici AL, Schiffer HH, Burstein ES, Lameh J, Mohell N, Hacksell U, Brann MR, Weiner DM (2004) Pharmacology of polymorphic variants of the human 5-HT1A receptor. Biochem Pharmacol 67(3):479–490. doi:10.1016/j.bcp.2003.09.030
Granas C, Nordvall G, Larhammar D (1998) Site-directed mutagenesis of the human 5-HT1B receptor. Eur J Pharmacol 349(2–3):367–375
Sherbiny FF, Schiedel AC, Maass A, Muller CE (2009) Homology modelling of the human adenosine A2B receptor based on X-ray structures of bovine rhodopsin, the beta2-adrenergic receptor and the human adenosine A2A receptor. J Comput Aided Mol Des 23(11):807–828. doi:10.1007/s10822-009-9299-7
Schiedel AC, Hinz S, Thimm D, Sherbiny F, Borrmann T, Maass A, Muller CE (2011) The four cysteine residues in the second extracellular loop of the human adenosine A(2B) receptor: role in ligand binding and receptor function. Biochem Pharmacol. doi:10.1016/j.bcp. 2011.05.008
Arseven OK, Wilkes WP, Jameson JL, Kopp P (2000) Substitutions of tyrosine 601 in the human thyrotropin receptor result in increase or loss of basal activation of the cyclic adenosine monophosphate pathway and disrupt coupling to Gq/11. Thyroid 10(1):3–10. doi:10.1089/10507250050137879
Sansuk K, Deupi X, Torrecillas I, Jongejan A, Nijmeijer S, Bakker R, Pardo L, Leurs R (2011) A structural insight into the reorientation of transmembrane domains 3 and 5 during family A GPCR activation. Mol Pharmacol 79(2):262–269. doi:10.1124/mol.110.066068
Acknowledgments
This research was performed under the auspices of the GPCR Forum, a program funded by the Dutch Top Institute Pharma (project D1-105). The authors thank Bas Vroling and the GPCRDB (http://www.gpcr.org/7tm/) for help with the cysteine occurrence analysis. They also are greatly indebted to Prof C.E. Mueller of Bonn University for the gift of [3H]PSB-603.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Peeters, M.C., Li, Q., van Westen, G.J.P. et al. Three “hotspots” important for adenosine A2B receptor activation: a mutational analysis of transmembrane domains 4 and 5 and the second extracellular loop. Purinergic Signalling 8, 23–38 (2012). https://doi.org/10.1007/s11302-011-9251-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-011-9251-x