
Abstract
Reduced representation genomic libraries (RRLs) are increasingly used to answer diverse questions in evolutionary biology, which remained unresolved otherwise. In the case of European beech (Fagus sylvatica L.), former applications of nuclear and chloroplast genetic markers indicated that most of the beech populations of Central, Eastern, and Northern Europe have a very homogeneous genetic structure. Thus, determining differentiation among populations of F. sylvatica at regional scale in Central Europe has remained a challenge for evolutionary biologists, mainly due to lack of variable genetic markers. In this study, we used the RAD-seq and GBS approaches to identify novel cpDNA polymorphisms that could be helpful to identify population structure of European beech at geographical scale. Our datasets allowed us to indicate a finer population structure and stronger patterns of spatial genetic structure than previous studies. It is expected that the applications of cpDNA-based phylogenetic approaches using whole-genome sequencing are able to provide even more detailed picture of species phylogeography than ever before.
Similar content being viewed by others


Introduction
During the last glacial period, European temperate forests were much more restricted than today and occupied mainly the southern European peninsulas, considered as main refugia (Bennett et al. 1991; Tzedakis et al. 2002). Actual spatial distribution of genetic diversity of forest tree species (but also other biota) is largely the result of the interplay between patterns of migration from refugia and the processes of adaptation to variable environmental conditions (Taberlet et al. 1998; Hewitt 1999).
Phylogeography is a powerful tool for inferring potential glacial refugia and post-glacial migration routes using spatial patterns of genetic polymorphism sampled from currently existing populations, as well as palaeodata (Hickerson et al. 2010). Many phylogeographic studies in angiosperms are based on chloroplast DNA polymorphisms for two main reasons. First, in the absence of DNA recombination during sexual reproduction and relatively low mutation rates, therefore, haplotypes remain mostly unchanged when passed to the next generation (King and Ferris 1998; Palmé 2002; Petit et al. 2002a, b). Second, chloroplast genome (cpDNA) is usually maternally inherited in angiosperms; therefore, cpDNA is dispersed through seeds and colonization patterns are not interfered by pollen flow (Rajora and Dancik 1992; Dumolin et al. 1995). However, deviations from strict maternal inheritance of cpDNA appear to be possible (McCauley et al. 2007), which may complicate the inference of phylogeographic structure. Knowledge of the historical distribution, postglacial migration, and expansion of a species is important to predict the potential fate of species in the future, especially under climate change conditions, as well as in conservation and management of its genetic resources (Zinck and Rajora 2016).
The European beech (Fagus sylvatica L.) is one of the most widespread species in deciduous forests in Europe (Leuschner et al. 2006; Packham et al. 2012). European beech contributes to both the high economic and ecological value through maintenance of European forest diversity and also provides diverse ecosystem services. For the first time, Demesure et al. (1996) studied phylogeography of beech using chloroplast DNA. They showed that a single haplotype predominates in a large part of the natural range of a species, but major parts of the range in the Apennines, Balkans, and Asia Minor were not included in that study. Nevertheless, it was proposed that the current distribution of beech in Central Europe derives from the post-glacial recolonization of populations originating mainly from the Balkan refugia (Demesure et al. 1996; Taberlet et al. 1998). In contrast, Magri et al. (2006), combining palaeo and genetic data, suggested that the main refugial areas, from where the recolonization to Central and Western Europe occurred, were located close to the Alps in Slovenia and south-western France and that the populations deriving from the Balkan Peninsula did not migrate to Northern Europe. Finally, both nuclear and chloroplast markers indicated that most of the beech populations of Central, Eastern, and Northern Europe have a very homogeneous genetic structure (Magri et al. 2006). Unfortunately, the available cpDNA markers did not detect differences among populations of F. sylvatica at a finer scale in Central Europe. On the other hand, considerable chloroplast diversity, geographically structured, was found in southern populations in Italy (Vettori et al. 2004) and in Greece (Hatziskakis et al. 2009; Papageorgiou et al. 2014). This raises the question if other cpDNA markers could be developed, which would contribute to a more detailed phylogeography of beech in Central Europe.
The increasing availability and affordability of next-generation DNA sequencing approaches (NGS) provides an impulse for researchers to switch from PCR-RFLP and microsatellite data analyses with relatively small numbers of loci to genome-wide sequencing methods that provide access to hundreds or thousands of loci for a number of individuals in a single run (Steele and Pires 2011). Now, sequencing of plastomes for hundreds of individuals is more easily achieved through whole-genome sequencing methods (Straub et al. 2012; Bock et al. 2014) or through targeted enrichment strategies (Mariac et al. 2014). With an increased number of chloroplast genomes sequenced, different studies showed many polymorphic sites when calling SNPs (Sabir et al. 2014). While the application of NGS methods for whole-genome population-scale studies may still be financially prohibitive, the chloroplast-based SNP markers identified in the representative sample of individuals using NGS methods may be further used to optimize SNP calling procedures targeting these loci in a future research.
In this study, we used two reduced representation libraries sequencing methods, i.e., restriction site-associated DNA sequencing (RAD-seq; Baird et al. 2008) and genotyping-by-sequencing (GBS; Elshire et al. 2011), to identify novel cpDNA polymorphisms in European beech, which would allow the detection of spatial genetic structure at a regional scale, particularly in Central Europe. For this purpose, we sampled 91 individuals from 47 European provenances, representing a wide geographical range of the species, with special attention to Central Europe. To detect novel chloroplast SNP markers, the DNA reads generated by the RAD-seq and GBS methods were subsequently aligned to the reference chloroplast genome of F. sylvatica. The identified SNP polymorphisms were tested for their nonrandom spatial distribution across the sampled area, discussing their usefulness in future phylogeographic studies.
Materials and methods
Plant material and DNA extraction
Samples of F. sylvatica were collected in April 2014 from one or two individuals representing 47 European provenances (91 individuals in total) growing in the common-garden experimental site located in Siemianice Experimental Forest District in south-central Poland (Fig. S1) (Barzdajn and Rzeznik 2002). Flushing leaf buds were sampled and dried at room temperature for 2 days. Desiccated tissue (30 mg) was cut into segments and placed in 2-ml Eppendorf tubes. Plates containing samples and 4-mm stainless steel grinding spheres were frozen and placed at − 20 °C overnight. Tissue samples were homogenized using grinding spheres in a mill (Mixer Mill MM 400, Retsch). DNA extraction was carried by using GeneMATRIXPlant & Fungi DNA Purification Kit (EURx, Poland), according to manufacturer’s protocol. DNA was quantified using the Eppendorf BioPhotometer (Eppendorf, Germany) and Quantus Fluorometer (Promega, Germany).
RAD-seq and GBS libraries preparation and sequencing
Restriction site-associated DNA sequencing (RAD-seq; Baird et al. 2008) and genotype-by-sequencing (GBS; Elshire et al. 2011) approaches were used to obtain single-nucleotide polymorphism (SNP) genetic markers. Extracted genomic DNA was normalized to a concentration of 30 ng/lL in 96-well plates and processed into the RAD-seq library using the restriction enzyme PstI, individually barcoded and sequenced by Floragenex Inc. (Eugene, OR, USA). The GBS library was prepared based on the ApeKI restriction enzyme, individually barcoded and sequenced by Cornell University, Biotechnology Resource Center (Ithaca, NY, USA). Libraries were sequenced in one lane run in 100-bp single-end mode on Illumina HiSeq2000 for both methods according to manufacturer’s instructions.
Quality control, demultiplexing, mapping, and SNP identification
Library quality and per-base quality were controlled with FastQC (Andrews 2010), and reads were checked for adaptor content, quality, quantity, and integrity of restriction enzyme cut sites. Raw sequence reads were demultiplexed based on the 10-bp barcodes in RAD-seq and 4–8 bp barcodes in GBS and were sorted individually using Stacks ver. 1.35 (Catchen et al. 2011). Only the reads with sufficiently high sequencing quality (Phred score ≥ 20) and with exact barcode match were retained for further analyses. The reads lengths were trimmed to 90 bp for the RAD-seq and to 64 bp for the GBS method to prevent false positives due to the decrease in base quality and adapter content towards the end of reads. Demultiplexed reads were subsequently aligned against the reference chloroplast genome of F. sylvatica (Vendramin et al., unpublished data) using BWA v.0.7.10 (Li 2013) with default parameters. Duplicate reads were removed using Picard (http://broadinstitute.github.io/picard/) and SAM files were converted to BAM files, sorted and indexed using SAMtools v.0.1.19 (Li et al. 2009). To identify SNP polymorphisms, we applied GATK v.3.3 (McKenna et al. 2010). Base quality score recalibration, indel realignment, SNP, and indel discovery and genotyping of all 91 samples were performed based on standard hard filtering parameters according to GATK Best Practices recommendations (DePristo et al. 2011; Van der Auwera et al. 2013). Only SNP polymorphisms with minimum depth of 10 reads, minor allele frequency (MAF) > 10%, and missing data per SNP < 25% were retained for further analyses.
Data analysis
To quantify the variation at chloroplast SNP loci, effective number of alleles and genetic diversity were calculated using FSTAT 2.9.3.2 software (Goudet 1995). The cpDNA was annotated using DOGMA (Dual Organellar GenoMe Annotator; Wyman et al. 2004). Haplotypes were determined as a combination of polymorphic SNP variants across the cpDNA loci. Haplotype Analysis ver. 1.05 software (Eliades and Eliades 2009) was used to estimate the following haplotype genetic diversity parameters: number of haplotypes (A), effective number of haplotypes (Ne), haplotypic richness (Rh; El Mousadik and Petit 1996), haplotypic diversity (H), and mean genetic distance between individuals (D2sh; Goldstein et al. 1995). Phylogenetic relationships among the established SNP haplotypes were depicted by a median-joining network analysis (Bandelt et al. 1999) within NETWORK ver 5.0.0.3 (available at http://www.fluxus-engineering.com/). Correlation between geographical and genetic distances between samples was tested based on Mantel tests (Mantel 1967) and Mantel’s correlograms, using the program PASSaGE ver.2 (Rosenberg and Anderson 2011). The significance of the Mantel test was evaluated based on 10,000 permutations.
Results
Genetic diversity
Mean number of sequence reads per individual passing QC filters was equal to 1.58 million for the RAD-seq method and 1.94 million for the GBS approach. Nearly 3.5% of the sequence reads were mapped to the cpDNA for RAD-seq and 1.32% for GBS. In order to improve the SNP discovery accuracy, we used only the uniquely mapped and non-redundant reads for downstream analyses. For the RAD-seq approach, 13 unique reads were mapped to chloroplast genome, with 6 SNPs detected. However, two pairs of SNPs appeared to be redundant and located within the same sequence reads; therefore, only a single SNP per read was selected for further analyses. After filtering, 94 sequence reads resulting from the GBS method were identified that mapped to cpDNA; however, among them, only 4 SNPs were identified. DNA sequences flanking the identified polymorphic sites are presented in Table S1. The mean coverage of the identified cpDNA SNPs (number of mapping sequence reads per individual) was 48 for the RAD-seq and 93.7 for the GBS data. We emphasize that sequence reads mapping to cpDNA still represents only a small fraction of the F. sylvatica chloroplast genome (size of 161,015 bp), as the sequence reads revealed 1.17 kb (0.73%) and 6.02 kb (3.74%)of the total cpDNA for RAD-seq and GBS, respectively. A total of 7 SNPs were identified in the regions of the four genes: psbA, psbD, rbcL, and atpA, and also 1 SNP was found in the intergenic region. The average genetic diversity across all 8 SNPs was 0.263 (Table 1).
Haplotypic diversity
The number of haplotypes identified was higher for the RAD-seq (7 haplotypes) than the GBS method (5 haplotypes). As a consequence, effective number of haplotypes, haplotype diversity, and mean genetic distance among individuals were all higher for the RAD-seq than that for the GBS approach (Table 2). However, the combination of alleles at all 8 polymorphic loci provided a set of 25 unique haplotypes. The haplotype diversity (H = 0.93) and haplotypic richness (Rh = 24) were high. The mean genetic distance between individuals was D2sh = 3.35 (Table 2). We constructed the median-joining networks among haplotypes defined based on RAD-seq, GBS, and a combination of both types of methods (Fig. S2). It appeared that the most frequent haplotypes were always located in the central parts of the network topologies indicating their ancestral character (Fig. S2).
Geographic distribution of chloroplast haplotypes
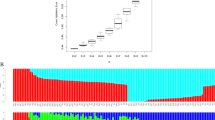
The geographic distribution of identified haplotypes suggests the presence of spatial structure of F. sylvatica in Central Europe (Fig. 1). Following the RAD-seq method, the most frequent haplotype H1 occurred widely across Europe, but was not present in Northern Europe (Fig. 1a). Haplotype H2 was present on the area from Denmark to south-eastern part of Poland; H3 appeared only in Central Europe; H4 and H5 occurred in several provenances originating from Poland and Germany, although H5 was also found in Slovakia. Haplotypes H6 and H7 were found mostly in Southern Europe.


Geographic distribution and frequency of chloroplast haplotypes in Fagus sylvatica based on RAD-seq (a), GBS (b), and combination of both types of markers (c)
In the case of the GBS method, the haplotype H1 appeared to be dominant and widespread over large parts of Europe; haplotype H2 was less frequent but it occurred on a large area from France, throughout Central Europe to Romania; haplotype H3 was particularly frequent near the border between Poland and Germany; finally, haplotypes H4 and H5 occurred mostly in Southeastern Europe, ranging from Slovakia to Romania (Fig. 1b).
Considering the most frequent haplotypes (n > 5) identified based on the combination of alleles obtained from both RRL methods (RAD-seq, GBS), their spatial distribution across Europe was also not random (Fig. 1c). Two of the 24 haplotypes (H1, H2) were found in more than 28% of the sampled individuals: H1 occurred in Northern and Southeastern Europe, while H2 was widespread over Central Europe from France throughout Germany to western Poland. Haplotypes H4 and H5 appeared in several provenances from Poland and Germany; H6 was found only in Poland and Slovakia; H7 occurred in Southern Europe and H8 was found only in Southwestern Europe. Rare haplotypes were mainly found in Southern Europe.
Spatial genetic structure
In general, correlations between genetic and geographic distances among sampled individuals appeared to be significant for the RAD-seq data (r = 0.083, p = 0.01), while non-significant for GBS (r = − 0.042) and combined datasets (r = 0.053), as revealed by Mantel test applied to all samples. However, when calculating Mantel r statistics for pairs of individuals within consecutive distance classes (often referred to as Mantel correlogram), significant positive correlations were found in the first three distance classes for RAD-seq and the combined dataset, and the first two distance classes for the GBS data (Table 3). Notably, the estimates of Mantel r statistics were higher for RAD-seq and the datasets generated based on both approaches jointly, than that for the GBS data alone, indicating stronger spatial genetic structure detected based on RAD-seq and combined datasets. In the latter case, the spatial autocorrelation appeared to be significant up to about 300 km.
Discussion
In recent years, cpDNA-based markers were efficiently used for phylogeographic studies in angiosperm forest tree species contributing to our understanding of their actual distribution and migratory routes since last glaciation (Demesure et al. 1996; Mousadik and Petit 1996; King and Ferris 1998; Dutech et al. 2000; Fineschi et al. 2000; Mohanty et al. 2001; Petit et al. 2002b; Heuertz et al. 2004; Bagnoli et al. 2015). However, inferences on phylogeography of Fagus sylvatica in Central Europe appeared to be difficult, mainly due to the lack of available polymorphic markers. In fact, in the previous study, Demesure et al. (1996) found only one PCR-RFLP chloroplast haplotype spanning across Central Europe, and similarly, based on cpDNA, Magri et al. (2006) identified only two PCR-RFLP haplotypes and just one microsatellite haplotype predominant in the same geographical areas.
In this paper, we found eight novel SNPs in the F. sylvatica chloroplast genome based on NGS sequencing of reduced representation genome libraries (RAD-seq and GBS methods), demonstrating that this approach can be an efficient way to identify novel SNPs in a chloroplast genome, once the reference cpDNA sequence is available. These novel markers provided considerable polymorphic information and indicated presence of spatial genetic structure across the central range of the species distribution, which was not possible with the already available markers (Demesure et al. 1996; Magri et al. 2006). Since Magri (2008) suggested that the recent postglacial expansion of F. sylvatica resulted from many small refugia, the availability of markers depicting geographic structure at a regional scale in Central Europe can be useful in defining routes and/or mechanisms of range expansion of this species. While the small sample size of this study prevents us from a detailed analyses and interpretation of phylogeographic structure, the identified haplotypes were not randomly distributed within the study area showing apparent spatial aggregation of haplotypes, on average up to about 250 km. Nevertheless, it might be expected that increasing the number of samples should provide a more detailed information about the routes and mechanisms of range expansion of F. sylvatica after the last glacial maximum towards central and northern parts of Europe.
For phylogeographic studies, SNPs may have some advantages over microsatellites or PCR-RFLP markers. First, SNPs are more densely and evenly distributed across the genome (Xing et al. 2005) and it has been shown that they display lower rates of genotyping errors (Montgomery et al. 2005). Moreover, SNPs are also less susceptible to homoplasy than microsatellites (Morin et al. 2009), which is important since homoplasy may affect the estimates of genetic distances (between individuals and populations), neighborhood size, and phylogenetic inference (Estoup et al. 2002). In particular, SNPs have an advantage over other types of genetic markers for the purposes of characterizing population divergence over long time scales.
We expect that the novel polymorphisms identified in this study will be useful for phylogeography studies in F. sylvatica. Although we provided DNA sequences flanking the identified SNPs (Table S1), these sequences are located within fairly conservative regions (7 SNPs located within genes) and can be used to design respective protocols for SNP genotyping. However, SNP genotyping by sequencing which was employed in this study is also possible. Other types of RRLs (such as ddRAD; Peterson et al. 2012) can be additional way for detecting further novel SNPs in the chloroplast genome. Ultimately, we anticipate that the resequencing of the full cpDNA genome of F. sylvatica at a larger number of individuals will provide more SNP data allowing for additional analyses of phylogeography of F. sylvatica in Europe based on chloroplast genome.
References
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 23 Dec 2018
Bagnoli F, Tsuda Y, Fineschi S, Bruschi P, González-Martínez S, Magri D, Zhelev P, Paule L, Simeone M, Vendramin GG (2015) Combining molecular and fossil data to infer demographic history of Quercus cerris: insights on European eastern glacial refugia. J Biogeogr 43:679–690. https://doi.org/10.1111/jbi.12673
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3(10):e3376. https://doi.org/10.1371/journal.pone.0003376
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barzdajn W, Rzeznik Z (2002) Wstepne wyniki miedzynarodowego doswiadczenia proweniencyjnego z bukiem [Fagus sylvatica L.] serii 1993/1995 w Lesnym Zakladzie Doswiadczalnym Siemnianice. Sylwan 146(02):149–164
Bennett KD, Tzedakis PC, Willis KJ (1991) Quaternary refugia of north European trees. J Biogeogr 18:103–115. https://doi.org/10.2307/2845248
Bock DG, Kane NC, Ebert DP, Rieseberg LH (2014) Genome skimming reveals the origin of the Jerusalem artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phytol 201(3):1021–1030. https://doi.org/10.1111/nph.12560
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short-read sequences. G3 (Bethesda) 1(3):171–182. https://doi.org/10.1534/g3.111.000240
Demesure B, Comps B, Petit RJ (1996) Chloroplast DNA phylogeography of the common beech (Fagus sylvatica L.) in Europe. Evolution 50(6):2515–2520. https://doi.org/10.1111/j.1558-5646.1996.tb03638.x
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Dally MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. https://doi.org/10.1038/ng.806
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91(8):1253–1256. https://doi.org/10.1007/BF00220937
Dutech C, Maggia L, Joly HI (2000) Chloroplast diversity in Vouacapoua americana (Caesalpiniaceae), a neotropical forest tree. Mol Ecol 9(9):1427–1432. https://doi.org/10.1046/j.1365-294x.2000.01027.x
Eliades NG, Eliades DG (2009) Haplotype analysis: software for analysis of haplotypes data. Distributed by the authors. Forest genetics and forest tree breeding. Georg-August University Goettingen, Goettingen. http://www.uni-goettingen.de/en/134935.html. Accessed 23 Dec 2018
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379. https://doi.org/10.1371/journal.pone.0019379
Estoup A, Jarne P, Cornuet JM (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11(9):1591–1604. https://doi.org/10.1046/j.1365-294X.2002.01576.x
Fineschi S, Taurchini D, Villani F, Vendramin GG (2000) Chloroplast DNA polymorphism reveals little geographical structure in Castanea sativa Mill. (Fagaceae) throughout southern European countries. Mol Ecol 9(10):1495–1503. https://doi.org/10.1046/j.1365-294x.2000.01029.x
Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139(1):463–471
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86(6):485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Hatziskakis S, Papageorgiou AC, Gailing O, Finkeldey R (2009) High chloroplast haplotype diversity in Greek populations of beech (Fagus sylvatica L.). Plant Biol 11(3):425–433. https://doi.org/10.1111/j.1438-8677.2008.00111.x
Heuertz M, Fineschi S, Anzidei M, Pastorelli R, Salvini D, Paule L, Frascaria-Lacoste N, Hardy OJ, Vekemans X, Vendramin GG (2004) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13(11):3437–3452. https://doi.org/10.1111/j.1365-294X.2004.02333.x
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68(1–2):87–112. https://doi.org/10.1111/j.1095-8312.1999.tb01160.x
Hickerson MJ, Carstens BC, Cavender-Bares J, Crandall KA, Graham CH, Johnson JB, Rissler L, Victoriano PF, Yoder AD (2010) Phylogeography’s past, present, and future: 10 years after. Mol Phylogenet Evol 54(1):291–301. https://doi.org/10.1016/j.ympev.2009.09.016
King RA, Ferris C (1998) Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol Ecol 7(9):1151–1161. https://doi.org/10.1046/j.1365-294x.1998.00432.x
Leuschner C, Meier IC, Hertel D (2006) On the niche breadth of Fagus sylvatica: soil nutrient status in 50 Central European beech stands on a broad range of bedrock types. Ann For Sci 63(4):355–368. https://doi.org/10.1051/forest:2006016
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv preprint arXiv 1303:3997
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Magri D (2008) Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J Biogeogr 35(3):450–463. https://doi.org/10.1111/j.1365-2699.2007.01803.x
Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latałowa M, Litt T, Paule L, Roure JM, Tantau I, Van Der Knaap WO, Petit RJ, De Beaulieu JL (2006) A new scenario for the quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytol 171(1):199–221. https://doi.org/10.1111/j.1469-8137.2006.01740.x
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27(2 Part 1):209–220
Mariac C, Scarcelli N, Pouzadou J, Barnaud A, Billot C, Faye A, Kougbeadjo A, Maillol V, Martin G, Sabot F, Santoni S, Vigouroux Y, Couvreur TLP (2014) Cost-effective enrichment hybridization capture of chloroplast genomes at deep multiplexing levels for population genetics and phylogeography studies. Mol Ecol Resour 14(6):1103–1113. https://doi.org/10.1111/1755-0998.12258
McCauley DE, Sundby AK, Bailey MF, Welch ME (2007) Inheritance of chloroplast DNA is not strictly maternal in Silene vulgaris (Caryophyllaceae): evidence from experimental crosses and natural populations. Am J Bot 94(8):1333–1337. https://doi.org/10.3732/ajb.94.8.13333
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. http://www.genome.org/cgi/doi/10.1101/gr.107524.110. Accessed 23 Dec 2018
Mohanty A, Martín JP, Aguinagalde I (2001) A population genetic analysis of chloroplast DNA in wild populations of Prunus avium L. in Europe. Heredity 87(4):421–427. https://doi.org/10.1046/j.1365-2540.2001.00922.x
Montgomery GW, Campbell MJ, Dickson P, Herbert S, Siemering K, Ewen-White KR, Visscher PM, Martin NG (2005) Estimation of the rate of SNP genotyping errors from DNA extracted from different tissues. Twin Res Hum Genet 8(4):346–352. https://doi.org/10.1375/twin.8.4.346
Morin PA, Leduc RG, Archer FI, Martien KK, Huebinger R, Bickham JW, Taylor BL (2009) Significant deviations from Hardy–Weinberg equilibrium caused by low levels of microsatellite genotyping errors. Mol Ecol Resour 9(2):498–504. https://doi.org/10.1111/j.1755-0998.2008.02502.x
Mousadik A, Petit RJ (1996) Chloroplast DNA phylogeography of the argan tree of Morocco. Mol Ecol 5(4):547–555. https://doi.org/10.1111/j.1365-294X.1996.tb00346.x
Packham JR, Thomas PA, Atkinson MD, Degen T (2012) Biological flora of the British Isles: Fagus sylvatica. J Ecol 100(6):1557–1608. https://doi.org/10.1111/j.1365-2745.2012.02017.x
Palmé AE (2002) Chloroplast DNA variation, postglacial recolonization and hybridization in hazel, Corylus avellana. Mol Ecol 11(9):1769–1779. https://doi.org/10.1046/j.1365-294X.2002.01581.x
Papageorgiou AC, Tsiripidis I, Mouratidis T, Hatziskakis S, Gailing O, Eliades NGH, Vidalis A, Drouzas AD, Finkeldey R (2014) Complex fine-scale phylogeographical patterns in a putative refugial region for Fagus sylvatica (Fagaceae). Biol J Linn Soc 174(4):516–528. https://doi.org/10.1111/boj.12148
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7(5):e37135. https://doi.org/10.1371/journal.pone.0037135
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam B, Deans JD, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Jensen JS, Konig AO, Lowe AJ, Kremer A (2002a) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manag 156(1):49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Petit RJ, Csaikl UM, Bordács S, Burg K, Coart E, Cottrell J, van Dam B, Deans JD, Dumolin- Lapegue S, Fineschi S, Finkeldey R, Gillies A, Glaz I, Goicoechea PG, Jensen JS, Konig AO, Lowe AJ, Madsen SF, Kremer A (2002b) Chloroplast DNA variation in European white oaks: phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag 156(1):5–26. https://doi.org/10.1016/S0378-1127(01)00645-4
Rajora OP, Dancik BP (1992) Chloroplast DNA inheritance in Populus. Theor Appl Genet 84(3):280–285. https://doi.org/10.1007/BF00229483
Rosenberg MS, Anderson CD (2011) PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol Evol 2(3):229–232. https://doi.org/10.1111/j.2041-210X.2010.00081.x
Sabir JS, Arasappan D, Bahieldin A, Abo-Aba S, Bafeel S, Zari TA, Edris S, Shokry AM, Gadalla NO, Ramadan AM, Atef A, Al-Kordy MA, El-Domyati FM, Jansen RK (2014) Whole mitochondrial and plastid genome SNP analysis of nine date palm cultivars reveals plastid heteroplasmy and close phylogenetic relationships among cultivars. PLoS One 9(4):e94158. https://doi.org/10.1371/journal.pone.0094158
Steele PR, Pires JC (2011) Biodiversity assessment: state-of-the-art techniques in phylogenomics and species identification. Am J Bot 98(3):415–425. https://doi.org/10.3732/ajb.1000296
Straub SC, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A (2012) Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot 99(2):349–364. https://doi.org/10.3732/ajb.1100335
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7(4):453–464. https://doi.org/10.1046/j.1365-294x.1998.00289.x
Tzedakis PC, Lawson IT, Frogley MR, Hewitt GM, Preece RC (2002) Buffered tree population changes in a quaternary refugium: evolutionary implications. Science 297(5589):2044–2047. https://doi.org/10.1126/science.1073083
Van der Auwera GA, Geraldine A, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics:11–10. https://doi.org/10.1002/0471250953.bi1110s43
Vettori C, Vendramin GG, Anzidei M, Pastorelli R, Paffetti D, Giannini R (2004) Geographic distribution of chloroplast variation in Italian populations of beech (Fagus sylvatica L.). Theor Appl Genet 109(1):1–9. https://doi.org/10.1007/s00122-004-1609-9
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17):3252–3255. https://doi.org/10.1093/bioinformatics/bth352
Xing C, Schumacher FR, Xing G, Lu Q, Wang T, Elston RC (2005) Comparison of microsatellites, single-nucleotide polymorphisms (SNPs) and composite markers derived from SNPs in linkage analysis. BMC Genet 6(1):S29. https://doi.org/10.1186/1471-2156-6-S1-S29
Zinck JW, Rajora OP (2016) Post-glacial phylogeography and evolution of a wide-ranging highly-exploited keystone forest tree, eastern white pine (Pinus strobus) in North America: single refugium, multiple routes. BMC Evol Biol 16(1):56. https://doi.org/10.1186/s12862-016-0624-1
Acknowledgements
We would like to thank Władysław Barzdajn from Poznań University of Life Sciences, Poland, for providing us samples for this study. We also thank Katarzyna Meyza and Ewa Sztupecka for the help in laboratory work.
Data archiving statement
The raw data of RAD-seq and GBS have been submitted to NCBI SRA database under the BioProject ID number PRJNA498382.
Funding
This study was supported by research grant from National Science Center, Poland (2012/04/A/NZ9/00500).
Author information
Authors and Affiliations
Contributions
The study was designed by J.M., B.U., and J.B.; B.U. collected samples and coordinated outsourced services; B.U. performed initial bioinformatics analyses and J.M. performed read mapping, SNP calling, and population genetic analyses; G.G.V. provided unpublished cpDNA sequence data; manuscript was written by J.M. with inputs and revisions provided by J.B., B.U., and G.G.V.; J.B. obtained funding.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by J. Wright
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1
Location of the sampled provenances (black dots) and natural distribution of Fagus sylvatica (green) (PNG 623 kb)
Fig. S2
Median-joining haplotype networks among haplotypes based on the RAD-seq (a), GBS (b) and combination of both types of methods (c). In the latter case, only haplotypes found in two (white circles) or more individuals are presented. Haplotype colors are the same as on Fig. 1. (DOCX 106 kb)
Table S1
DNA sequences flanking SNPs in Fagus sylvatica; polymorphic site with nucleotide variants is indicated within parentheses (major/minor allele) (DOC 37 kb)
Table S2
SNP genotypes of individuals (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Meger, J., Ulaszewski, B., Vendramin, G.G. et al. Using reduced representation libraries sequencing methods to identify cpDNA polymorphisms in European beech (Fagus sylvatica L). Tree Genetics & Genomes 15, 7 (2019). https://doi.org/10.1007/s11295-018-1313-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-018-1313-6