
Abstract
Functional genomics has made possible advanced structure-to-function investigation of pathogens and helped characterize virulence mechanisms. Proteomics has been become a tool for large-scale identification of proteins involved during invasion and infection by the pathogens. Bacterial surface and secreted proteins play key role in the interaction between the bacterial cell and the host environment. Thus exoproteome and surface proteome of a microorganism are hypothesized to contain components of effective vaccines. Surfome and exoproteome analysis strategy facilitates identification of novel vaccine antigen and overall helps in progress of discovery of vaccine. The study of the antibody response can advance how proteomics is used, because it investigates antibody–antigen interactions and also unravel the relationship of antibody responses to pathogen and host characteristics. System immunology integrating with proteome i.e. immunoproteomics is applicable to those infections that are having tendency of diverse antibody target recognition and thus accurately reflects progression of the infection.
Similar content being viewed by others


Introduction
Proteins of any pathogen that are displayed on the cell surface, membrane bound or secreted into the extracellular surrounding are primarily required for cell- cell contact between the host and the pathogen. Accessibility to the genomic sequence of a pathogen has disclosed its entire antigenic repertoire. Sequence-based ‘Reverse Vaccinology’ approaches (Rappuoli and Covacci 2003) have catalysed a major drift in development of vaccine. This approach screens entire pathogen’s genome by high-throughput in silico method and identifies targets with vaccine attributes.
Proteomic technologies (cataloguing the entire protein content of a cell) serve as an important complement to the reverse vaccinology approach to antigen discovery (Dove 1999). Advancement in protein separation technologies, in form of combination with mass spectrometry and genome sequencing, has made feasible the task of elucidation of total protein components (proteome) of any pathogen at its best. The most popular procedure in proteome analysis is 2D-gel electrophoresis in which the protein mixture is first resolved into its individual components. However, for in depth analysis and identification of low abundance proteins, gel-free separation methodologies are becoming more popular. For determination of accurate molecular masses using mass spectrometry, each protein is digested with a specific protease to generate discrete peptide fragments (Wöhlbrand et al. 2013). Matrix-Assisted Laser Desorption/Ionization (MALDI) and Electro Spray Ionization (ESI) are generally used as ion source for soft ionization of peptide molecules. Time of flight mass spectrometer (MALDI-TOF MS) or other formats are being extensively used for the comparison of the experimentally generated peptide-mass fingerprint with the in silico-generated theoretical fingerprints using a database of all proteins predicted by genome sequencing (Grandi 2001). Fragmentation of peptide precursors in tandem mass spectrometry further provides the sequence information leading to unambiguous identification of proteins and beyond.
Pathogens tend to use multiple proteins during host infection against host defence. Proteomic techniques enable identification of those proteins and their subset proteins residing on the surface of the pathogen (Walters and Mobley 2010). Vaccine targets once identified from a pathogen’s proteome can be expressed as recombinant proteins and then further tested for their immunogenicity in appropriate in vivo model (Seib et al. 2012).
Proteomics and vaccine development
Proteomics permits the study of the virulence mechanisms in pathogen on mass- scale and become a powerful tool for high-throughput analysis of proteins. It not only captures a metabolic snapshot at a particular moment in the life of a pathogen, but also provides the whole information of a protein like its localization, function, characteristic and interaction with host proteins. Comparative proteomics can expose important information on the differences between attenuated and pathogenic organisms and whether a protein is conserved among various strains (DelVecchio et al. 2010). Nowadays, proteomics is combined with two relatively emerging areas: immunomics (looking for immunogenic proteins) and vaccinomics (characterization of host response to immunization), that yields valuable information on host- pathogen interaction. It also gives a pace to the process of the identification and detailed characterization of novel antigens, having vaccine potential (Adamczyk-Poplawska et al. 2011).
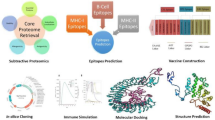
Over the past decade, proteomic approaches combining 2-DE and MS have been used to systematically map the cellular, surface-associated and secreted proteins of many pathogens. Availability of complete genomic sequences and the amalgamation of proteomic, genomic and bioinformatic technologies have facilitated the identification of potential vaccine candidate and therapeutic agents (Fig. 1). 2DE proteome analysis has been performed for several bacteria and comprehensive and comparative results have been published for several bacterial species. Expression proteomic studies and elucidation of reference maps for one or more of sub-cellular fractions from cytosolic, membrane, cell surface and extra cellular proteome have been carried out for several pathogenic bacteria including Staphylococcus aureus (Cordwell et al. 2002), Bacteroides fragilis (Diniz et al. 2004), Aeromonas salmonocida (Ebanks et al. 2005), Helicobacter pylori (Backert et al. 2005), Mycobacterium tuberculosis (Mattow et al. 2003), Escherichia coli (Molloy et al. 2000), Bacillus anthracis (Huang et al. 2004), and Chlamydia pneumoniae (Molestina et al. 2002).

Schematic overview of the way in which proteomics, bioinformatics and associated high-throughput analyses applied to understand pathogen and its interactions and discover vaccine candidates
Bioinformatic selection of candidate vaccine antigen coupled with proteomic identification
Whilst empirical approaches to the selection of vaccine sub-units are still employed, bioinformatics approaches to select candidate protein sub-units from bacterial genome sequences have been used recently in the last decade. The computer aided discovery of protein antigen i.e. immunoinformatics has significantly reduced the number of laboratory tests and resources required for identification of candidate vaccine antigen. These all in silico approaches including the mining of genome sequence are cumulatively termed as reverse vaccinology (Rappuoli and Covacci 2003). Reverese vaccinology has been applied for vaccine development and a few examples in this regard include Group B meningococcus (MenB), M. tuberculosis (Mattow et al. 2003; Tsolakos et al. 2014).
Generally ‘in silico’ approaches to the identification of vaccine antigens have relied on these criteria for selection of candidate protein: (a) prediction of sub-cellular location (b) identification of antigens using sequence similarity search and (c) the use of sophisticated statistical approaches for predicting the probability of antigen characteristics.
The reductive strategy was used in B. anthracis for identification of surface antigen. The selection procedure contained combination of preliminary annotation (sequence similarity searches and domain assignments) and some additional filtering criteria like prediction of cellular localization, taxonomical and functional screen, and number of paralogs. Preliminary screening by in silico method was followed by proteomic analysis of B. anthracis membrane fraction (Ariel et al. 2003). On the basis of theory of reverse vaccinology, bioinformatics study, comparative genomic hybridization (CGH) analysis and transcriptional analysis were carried out to identify vaccine candidate in Leptospira interrogans to diagnose leptospirosis (Yang et al. 2006). Comparative genomics along with bioinformatics approach was used for identification of novel vaccine candidate in Enterohemorrhagic E. coli (García-Angulo et al. 2014).
Vaxign (He et al. 2010) and NERVE (Vivona et al. 2006) are two tools that predict vaccine candidate and protective antigen based on reverse vaccinology approach. A multi-step antigen selection approach was used initially by applying Vaxign tool to predict ORF encoding outer membrane proteins with antigenic determinants and then in silico prediction for down-selection of the candidate antigens (Gomez et al. 2013). In one of the study, five complete Corynebacterium pseudotuberculosis genomes were subjected to pan-genomic analysis to reveal in silico predicted pan- exoproteome which further helped in generating a list of putative vaccine candidate (Santos et al. 2012). Thus, vaccine informatics is emerging as a current research area that facilitates rational vaccine design using immunoinformatics and epitope prediction tool.
Exoproteome analysis for virulence determinants and vaccine discovery
As in any living cell, extracytoplasmic proteins play versatile functions, including nutrient uptake, chemo-sensing, motility, adherence, and impact on host cells. Moreover, they deliver distinguishing clues regarding the physiology and bacterial cell interaction in an ecological niche.
A pathogen must be able to evade the host immune system in order to invade, adhere, and colonisation of host cells. The factors involved in these processes are also of great importance for the understanding of pathogenesis but unfortunately they are largely unknown. The exoproteins of Gram-positive bacteria are likely to contain some of these key factors. The protein secretion systems and the secreted proteins collectively account for Secretome; in monoderm bacteria (Gram-positive cell envelope architecture), these proteins can also be found in the membrane and/or cell wall. Hence the proteins found in the extracellular milieu of Gram-positive bacteria are called extracellular proteins, or exoproteins, which constitute exoproteome; not necessarily secreted by known secretion systems.
C. perfringens exoproteome was elucidated and several proteins were identified in the exoproteome of C. perfringens type A and type C strains. Some of those exoproteins had features characteristic of virulence determinants, suggesting that in addition to the “classic” extracellular toxins, a large number of proteins may be essential for C. perfringens virulence. One of the candidate Ornithine carbamoyltransferase was also shown to be protective in mice and thus making it a potential vaccine candidate. The investigation demonstrated the strength of proteomics in the rapid elucidation of vaccine candidates and was the turning point for the identification of diagnostic markers for C. perfringens and the development of vaccines against gas gangrene in humans (Sengupta et al. 2010). Recently, from the exoproteome of C. perfringens, three putative virulence associated lipoproteins; polysaccharide deacetylase family protein, probable ion-uptake ABC transporter, and a putative lipoprotein of no known function were reported with respect to their immuno-protective potentials. The three proteins were over expressed and purified to near homogeneity and found to generate protective immune response in mice against challenge with C. perfringens (Dwivedi et al. 2015a).
The secretome of the three C. difficile strains were analyzed and compared. Proteins were separated and analyzed by means of SDS-PAGE and LC–MS. LC–MS analysis revealed 158 different proteins in the supernatant of C. difficile. Most of the identified proteins originated from the cytoplasm (Boetzkes et al. 2012). Exoproteome of another Gram positive pathogen Listeria monocytogenes was presented using amalgam of bioinformatic and experimental proteomic approaches. Proteomic analysis using both 2D gel electrophoresis/MALDI and HPLC/ESI–MS identified 105 proteins in the culture supernatant of L. monocytogenes. Among these, already known virulence determinant were detected, showing the importance of this sub-proteome. Comparative proteomic analysis between pathogenic and non-pathogenic species of Listeria was carried out to uncover proteins probably involved in pathogenicity and/or adaptation in the host (Trost et al. 2005).
Similarly, several virulence markers secreted by Streptococcus pyogenes are involved in pathogenesis. One of the important factors is peroxide regulator (PerR) which is associated with the peroxide resistance response and pathogenesis. Very little information about the regulation of PerR and its involvement in virulence is known so far. Comparative secretome analysis of a perR deficient mutant with a wild-type strain investigated the role of PerR in regulating the expression of the S. pyogenes secretome involved in virulence (Wen et al. 2011). Secretome at different time course of B. anthracis was investigated to analyze pathogenicity progression, identify therapeutics and diagnostic markers, and for the elucidation of vaccine targets (Walz et al. 2007). Moreover, secretory proteins of human pathogen Burkholderia cepacia are known to be involved in virulence and participate in host- pathogen interaction. Secretome of B. cepacia identified eighteen immunogenic proteins and might be potential vaccine candidate (Mariappan et al. 2010). More recently, exoproteome and secretome approach was employed in pathogen H. pylori to identify potential candidate vaccine for the development of multicomponent vaccine and it is also detected that exoproteome composition of the H. pylori is also growth phase dependent (Naz et al. 2015; Snider et al. 2016).
Pacheco et al. (2011) identified 93 different C. pseudotuberculosis extracellular proteins by analyzing the exoproteomes of two strains isolated from different hosts pertaining distinct virulence phenotypes under in vitro conditions. In another study, L. interrogans serovar Lai was grown in proteinfree medium to obtain its set of extracellular proteins for analysis by LC/MS. Many virulence factors were detected in the supernatant, providing new insights into the pathogenesis of leptospirosis (Zeng et al. 2013).
Surface proteome analysis for virulence determinants and vaccine discovery
Bacterial surface proteins play a fundamental role in the interaction between the bacterial cell and its environment. Surface proteins contribute to escape from the host immune attack, to biofilm formation and in mounting defence against host response. The ability of surface proteins to bind to host extracellular matrix and plasma components promotes adhesion to host tissues and invasion of cells. Hence, surface proteins are good targets of drugs while aiming towards prevention of bacterial infections and diseases. Furthermore, because surface proteins directly interact with the host immune system, they are likely to become components of effective vaccines. Vaccines based on surface-exposed and secreted proteins for some pathogens are already commercially available and others are in development.
Comparative genomic analysis of two closely related species of Clostridium pathogen-C. tetani and C. perfringens suggested many homologous ORFs in both of the genomes. This striking finding suggested an extensive sharing of common epitopes between homologous proteins of these two medically important pathogens. In one study, total cellular proteins of C. tetani were probed with antisera raised against whole cells of C. perfringens ATCC13124 (Alam et al. 2008). Cross-reactive proteins were identified by mass spectrometry. Immuno-fluorescence microscopy indicated binding of the polyclonal C. perfringens whole cell antibody to the surface of both C. perfringens and C. tetani, indicating possibility of some of the cross reactive proteins to be actually surface localized. Also, the finding provided basis for the search of potential vaccine candidates with broader coverage, encompassing more than one pathogenic clostridial species. In another report, predominant cell surface and cell envelope (structure associated) proteins were identified from C. perfringens ATCC13124 differentially expressed in cells grown on cooked meat medium (CMM) in comparison with cells grown in reference state (tryptose-yeast extract-glucose medium). Two of the surface proteins of C. perfringens, phosphoglycerate kinase and ornithine carbamoyltransferase were found immunogenic and later one was reported as protective in animal model. Three proteins phosphoglycerate kinase, N-acetylmuramoyl-l-alanine amidase, and translation elongation factor Tu and EF-G were predicted to be putative vaccine candidates on the basis of their high abundance on the cell surface and their homologs in other Gram positive pathogenic bacteria were previously shown to be immunogenic and/or virulence determinants (Alam et al. 2009). Recently, extractable proteome of total four strains from type A and type C (two strains from each type) identified a total of 134 unique proteins (529 protein spot features). Proteins showing altered expression under host-simulated conditions from virulent type A strain (ATCC13124) were also elucidated which provides a starting point to explore their potentials in detection/diagnostic and there possible role in pathogenesis of the organism (Dwivedi et al. 2015b).
Cell wall proteins from C. difficile were extracted employing two methods of extraction: (1) low pH glycine treatment and (2) lysozyme digestion of the cell wall, and analysed in detail by 2DGE and mass spectrometry. Several proteins were identified as paralogs of the surface layer proteins (SLPs), consistent with the hypothesis that these are cell wall associated proteins (Wright et al. 2005). Moreover, cell wall fraction from Streptococcus pneumoniae was extracted and approximately 150 soluble proteins were identified by 2D gel electrophoresis. Two of them were cross-reactive with unrelated strains of different serotypes and conferred protection against respiratory challenge with virulent pneumococci (Ling et al. 2004). Five potential candidate proteins from surface fractions of 40 different serotypes of S. pneumoniae were screened for inclusion in a vaccine (Morsczeck et al. 2008).
One rapid and more direct method for the “in situ” identification of surface proteins is “shaving” the surface of intact bacteria with proteolytic enzymes, followed by identification of the released peptides by liquid chromatography (LC) and tandem mass spectrometry (MS/MS) (Rodríguez-Ortega et al. 2006). An advantage of this approach is limited contamination with intracellular proteins because the proteolytic enzymes will only have access to proteins that are exposed on the surface of the bacterial cell. “Shaving” approach is applied to identify the surface proteome of E. faecalis V583 (Bøhle et al. 2011), Group A Streptococcus (Rodríguez-Ortega et al. 2006), Group B streptococcus (Doro et al. 2009), Uropathogenic E. coli (Walters and Mobley 2009) and S. pneumoniae (Mattow et al. 2003).
Surface-exposed proteins of complete set of group A Streptococcus (GAS) were characterized showing majority of the identified proteins belonging to families of predicted surface-associated proteins, and found to be immunogenic when polyclonal antibodies were raised against the corresponding recombinant proteins. Most of them were described as protective antigens in the available literature and one new antigen, capable of conferring consistent protection in the mouse against challenge with a virulent GAS strain was also discovered (Rodríguez-Ortega et al. 2006). When this strategy was applied to the Group B Streptococcus, 43 surface-associated proteins were identified, including all protective antigens. Using a similar approach, Bøhle et al. (2011) revealed 69 surface-located proteins in E. faecalis V583 with varying known or conceivable functions in bacterial cell. The authors also found few proteins with unknown functions. Several identified proteins were involved in cell wall synthesis and maintenance as well as in cell–cell communication and were hypothesised as putative targets for drug design. In Uropathogenic E. coli (UPEC) two approaches was used to elucidate surface proteins; One method was shaving of surface exposed peptides and other was the labelling of whole bacterial cell with a biotin tag followed by two-dimensional liquid chromatography-tandem mass spectrometry (2-DLC-MS/MS). These methods identified 25 virulence proteins, of which one was appeared to be a possible effective vaccine candidate (Walters and Mobley 2010).
In other study, Surface proteome of M. avium subsp. hominissuis was characterized using: selective biotinylation of surface-exposed proteins, streptavidin affinity purification, and shotgun mass spectrometry. Surface proteins unique to the disease were identified in specific culture conditions mimicking early infection. That proteomic report facilitated better understanding towards establishment of M. avium subsp. hominissuis infection (McNamara et al. 2012). More recently, in vitro core surface proteome of Mycoplasma mycoides subsp. Mycoides was characterized that will certainly facilitate prioritization of candidate Mycoplasma antigens for improved control measures, surface-exposed membrane proteins will include those that are involved in host-pathogen interactions (Krasteva et al. 2014). Proteomic analysis of surface immunogens from serogroup B Neisseria meningitidis also carried out for inclusion in subunit vaccines against bacterial meningitis and discovery of vaccine candidates against other bacterial infections (Tsolakos et al. 2014). More recently, to identify potential gonococcal vaccine antigens, cell envelope proteins of N. gonorrhoeae expressed under different environmental stimuli were elucidated using comprehensive proteomic platform (Zielke et al. 2016).
How surface proteins are targeted to the cell surface while crossing the membrane and thick peptidoglycan and remain associated there, are some burning issues for improved understanding towards clinical relevance of these proteins. Certainly, in depth analysis of molecular sorting mechanisms may give idea to identify new therapeutic targets for Gram positive bacteria. Moreover, proteins exposed to surface are prime targets for the host immune system, and thus their identification can aids to the development of novel treatments, and increase the repertoire of vaccines for wide range of disease.
Immunoproteomics for virulence determinants and vaccine discovery
Investigation and evaluation of the host-pathogen response are two essential aspects to vaccine development. Elicitation of cell mediated immune response either driven by Th1 in case of intracellular pathogen or Th2 in extracellular pathogen is strongly required for protection against any pathogen. These all consequences enhance antibody production and antibody-mediated cell killing. Additionally, in vitro knowledge of the interactions between pathogens and host cells is also crucial for investigation of bacterial components that attach to host cells.
Immunoproteomics is an extension of proteomics, which permits specific elucidation of antigens based on immunoreactivity. In process of immunoproteomics, 2-D blots are probed with serum collected from host post infection. This process has bypassed the lengthy process of testing of immunoreactivity and hence vastly boosted the vaccine discovery by directly allowing the identification of those novel proteins which evoke immune system.
The natural step up to, whereby 2-D blots are probed with host serum following infection or immunisation has greatly enhanced the identification of potential vaccine candidates, by enabling the discovery of novel proteins that stimulate the humoral host immune system. The main advantage of this approach is that bacterial proteins have already been processed and modified post-translationally in host system so finally expressed protein is obtained for analysis. Several groups have explored various potential vaccine candidates using this approach which is discussed below.
In S. pneumoniae immunoproteomics has been used to identify immunogenic proteins. 2-D blot of cell wall fractions from S. pneumoniae was probed with sera from healthy children or adults ravelled seventeen immunoreactive proteins. Of these, two proteins were further found to be protective in a mouse challenge model (Mizrachi Nebenzahl et al. 2007).
Furthermore, immunoproteomic analysis of the S. pneumoniae secretome also identified several novel antigens which were shown to be immunogenic (Choi et al. 2012). In another report, the use of immunoproteomics was to visualize and identify immunogenic Shigella flexneri soluble and membrane proteins, which were reactive to sera from S. flexneri infected patients (Jennison et al. 2006).
Another example of the immunoproteomic approach is, in identification of novel vaccine candidates from N. meningitidis. In that study one immunoreactive protein was evaluated as vaccine component after getting protection data from mice (Hsu et al. 2008). In another study, numerous candidate proteins were elucidated from the immunome of Brucella abortus cell envelope for discovery of vaccines against Brucellosis in cattle and humans (Connolly et al. 2006) Furthermore, immunogenic proteins from soluble fraction of Brucella melitensis 16M and M5 were identified using an immunoproteomic assay for developing subunit vaccine against Brucella infection (Yang et al. 2011; Zhao et al. 2011).
In an immunoproteomics study of piscine Streptococcus agalactiae cellular proteins, four novel immunoreactive proteins were implicated as vaccine candidates and virulence factors (Liu et al. 2013). Pang and coworkers (2010) reported first immunoproteomic approach in Vibrio to identify immunogenic proteins of V. harveyi, which provides a valuable tool for developing the protective antigens in future. There is an urgent need to develop polyvalent vaccine candidates that can protect from a broad spectrum of pathogen. For this, heterogeneous antiserum-based immunoproteomics approaches have been employed to explore outer membrane proteins with similar antigenicity that could be used as a cross-protective vaccine against several species of Vibrio (Li et al. 2010). More recently, a diverse set of antigenic proteins, were identified by analysing conidial and hyphal immunomes of the filamentous pathogenic fungus Lomentospora prolificans against healthy human serum which may be further used to discover potential therapeutic targets (Pellon et al. 2016).
Immunoproteomics approach has also been applied to combat viral diseases. To develop a universal flu vaccine, it has been implemented for the direct identification of HLA class I presented epitopes in the last decade, and recognized as a method for the identification of T cell epitopes. In one study, combination of T cell epitopes specifically occur on influenza A-infected cells and a cross-reactive epitope displayed by the ectodomain of influenza M2, was used to evaluate T cell immunity in influenza infection and uncover universal antigen against it (Testa et al. 2012).
Collectively, exposure of bacterial cell proteins to the host immune system provides a pool of the proteins having vaccine candidate potential.
Conclusion
Omics approaches have been used for the lengthy process of identification of subunit vaccine and candidate protein vaccine antigens. As proteins presented on the cell surface or secreted by pathogen are first encountered in host cell. Thus detailed investigation of surface proteome and extracellular proteome of a pathogen may open up a significant opportunity to elucidate proteins associated with disease pathogenesis or having potential as surface markers. A protein antigen capable of neutralizing immune response results into most efficient subunit vaccine. When proteomics is coupled to western blotting, namely immunoproteomics, much of the information about immunogenic proteins can be derived accurately. Detection of immunoreactive antigens using serum highlights immunogenic proteins that are expressed during infection. Thus, the immunoproteomics based approach also appears to be a suitable tool for identification of novel vaccine candidates. The approaches discussed herein can lead to a deeper knowledge of the mechanisms underlying bacterial invasion, colonization, and pathogenesis.
References
Adamczyk-Poplawska M, Markowicz S, Jagusztyn-Krynicka EK (2011) Proteomics for development of vaccine. J Proteom 74:2596–2616
Alam SI, Bansod S, Singh L (2008) Immunization against Clostridium perfringens cells elicits protection against Clostridium tetani in mouse model: identification of cross-reactive proteins using proteomic methodologies. BMC Microbiol 8:194
Alam SI, Bansod S, Kumar RB, Sengupta N, Singh L (2009) Differential proteomic analysis of Clostridium perfringens ATCC13124; identification of dominant, surface and structure associated proteins. BMC Microbiol 9:162
Ariel N, Zvi A, Makarova KS, Chitlaru T, Elhanany E, Velan B, Cohen S, Friedlander AM, Shafferman A (2003) Genome-based bioinformatic selection of chromosomal Bacillus anthracis putative vaccine candidates coupled with proteomic identification of surface-associated antigens. Infect Immun 71(8):4563–4579
Backert S, Kwok T, Schmid M, Selbach M, Moese S, Peek RM Jr, König W, Meyer TF, Jungblut PR (2005) Subproteomes of soluble and structure-bound Helicobacter pylori proteins analyzed by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 5:1331–1345
Boetzkes A, Felkel KW, Zeiser J, Jochim N, Just I, Pich A (2012) Secretome analysis of Clostridium difficile strains. Arch Microbiol 194:675–687
Bøhle LA, Riaz T, Egge-Jacobsen W, Skaugen M, Busk ØL, Eijsink VG, Mathiesen G (2011) Identification of surface proteins in Enterococcus faecalis V583. BMC Genom 12:135
Choi CW, Lee YG, Kwon SO, Kim HY, Lee JC, Chung YH, Yun CY, Kim SI (2012) Analysis of Streptococcus pneumoniae secreted antigens by immuno-proteomic approach. Diagn Microbiol Infect Dis 72:318–327
Connolly JP, Comerci D, Alefantis TG, Walz A, Quan M, Chafin R, Grewal P, Mujer CV, Ugalde RA, DelVecchio VG (2006) Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 6(13):3767–3780
Cordwell SJ, Larsen MR, Cole RT, Walsh BJ (2002) Comparative proteomics of Staphylococcus aureus and the response of methicillin-resistant and methicillin-sensitive strains to Triton X-100. Microbiology 148:2765–2781
DelVecchio VG, Sabato MA, Trichilo J, Dake C, Grewal P, Alefantis T (2010) Proteomics for the development of vaccines and therapeutics. Crit Rev Immunol 30:239–254
Diniz CG, Farias LM, Carvalho MA, Rocha ER, Smith CJ (2004) Differential gene expression in a Bacteroides fragilis metronidazole-resistant mutant. J Antimicrob Chemother 54:100–108
Doro F, Liberatori S, Rodríguez-Ortega MJ, Rinaudo CD, Rosini R, Mora M, Scarselli M, Altindis E, D’Aurizio R, Stella M, Margarit I, Maione D, Telford JL, Norais N, Grandi G (2009) Surfome analysis as a fast track to vaccine discovery: identification of a novel protective antigen for Group B Streptococcus hypervirulent strain COH1. Mol Cell Proteom 8:1728–1737
Dove A (1999) Proteomics: translating genomics into products? Nat Biotechnol 17:233–236
Dwivedi P, Alam SI, Kumar O, Kumar RB (2015a) Lipoproteins from Clostridium perfringens and their protective efficacy in mouse model. Infect Genet Evol 34:434–443
Dwivedi P, Alam SI, Kumar O, Kumar RB (2015b) Comparative analysis of extractable proteins from Clostridium perfringens type A and type C strains showing varying degree of virulence. Anaerobe 35(Pt B):77–91
Ebanks RO, Goguen M, McKinnon S, Pinto DM, Ross NW (2005) Identification of the major outer membrane proteins of Aeromonas salmonicida. Dis Aquat Organ 68:29–38
García-Angulo VA, Kalita A, Kalita M, Lozano L, Torres AG (2014) Comparative genomics and immunoinformatics approach for the identification of vaccine candidates for enterohemorrhagic Escherichia coli O157:H7. Infect Immun 82(5):2016–2026
Gomez G, Pei J, Mwangi W, Adams LG, Rice-Ficht A, Ficht TA (2013) Immunogenic and invasive properties of Brucella melitensis 16 M outer membrane protein vaccine candidates identified via a reverse vaccinology approach. PLoS One 8(3):e59751
Grandi G (2001) Antibacterial vaccine design using genomics and proteomics. Trends Biotechnol 19:181–188
He Y, Xiang Z, Mobley HL (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol 2010:297505
Hsu CA, Lin WR, Li JC, Liu YL, Tseng YT, Chang CM, Lee YS, Yang CY (2008) Immunoproteomic identification of the hypothetical protein NMB1468 as a novel lipoprotein ubiquitous in Neisseria meningitidis with vaccine potential. Proteomics 8(10):2115–2125
Huang CM, Elmets CA, Tang DC, Li F, Yusuf N (2004) Proteomics reveals that proteins expressed during the early stage of Bacillus anthracis infection are potential targets for the development of vaccines and drugs. Genom Proteoms Bioinform 2:143–151
Jennison AV, Raqib R, Verma NK (2006) Immunoproteome analysis of soluble and membrane proteins of Shigella flexneri 2457T. World J Gastroenterol 12(41):6683–6688
Krasteva I, Liljander A, Fischer A, Smith DG, Inglis NF, Scacchia M, Pini A, Jores J, Sacchini F (2014) Characterization of the in vitro core surface proteome of Mycoplasma mycoides subsp. mycoides, the causative agent of contagious bovine pleuropneumonia. Vet Microbiol 168(1):116–123
Li H, Ye MZ, Peng B, Wu HK, Xu CX, Xiong XP, Wang C, Wang SY, Peng XX (2010) Immunoproteomic identification of polyvalent vaccine candidates from Vibrio parahaemolyticus outer membrane proteins. J Proteome Res 9(5):2573–2583
Ling E, Feldman G, Portnoi M, Dagan R, Overweg K, Mulholland F, Chalifa-Caspi V, Wells J, Mizrachi-Nebenzahl Y (2004) Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin Exp Immunol 138(2):290–298
Liu G, Zhang W, Lu C (2013) Identification of immunoreactive proteins of Streptococcus agalactiae isolated from cultured tilapia in China. Pathog Dis 69(3):223–231
Mariappan V, Vellasamy KM, Thimma JS, Hashim OH, Vadivelu J (2010) Identification of immunogenic proteins from Burkholderia cepacia secretome using proteomic analysis. Vaccine 28(5):1318–1324
Mattow J, Schaible UE, Schmidt F, Hagens K, Siejak F, Brestrich G, Haeselbarth G, Müller EC, Jungblut PR, Kaufmann SH (2003) Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24:3405–3420
McNamara M, Tzeng SC, Maier C, Zhang L, Bermudez LE (2012) Surface proteome of “Mycobacterium avium subsp. hominissuis” during the early stages of macrophage infection. Infect Immun 80(5):1868–1880
Mizrachi Nebenzahl Y, Bernstein A, Portnoi M, Shagan M, Rom S, Porgador A, Dagan R (2007) Streptococcus pneumoniae surface-exposed glutamyl trna synthetase, a putative adhesin, is able to induce a partially protective immune response in mice. J Infect Dis 196:945–953
Molestina RE, Klein JB, Miller RD, Pierce WH, Ramirez JA, Summersgill JT (2002) Proteomic analysis of differentially expressed Chlamydia pneumoniae genes during persistent infection of HEp-2 cells. Infect Immun 70:2976–2981
Molloy MP, Herbert BR, Slade MB, Rabilloud T, Nouwens AS, Williams KL, Gooley AA (2000) Proteomic analysis of the Escherichia coli outer membrane. Eur J Biochem 267:2871–2881
Morsczeck C, Prokhorova T, Sigh J, Pfeiffer M, Bille-Nielsen M, Petersen J, Boysen A, Kofoed T, Frimodt-Møller N, Nyborg-Nielsen P, Schrotz-King P (2008) Streptococcus pneumoniae: proteomics of surface proteins for vaccine development. Clin Microbiol Infect 14:74–81
Naz A, Awan FM, Obaid A, Muhammad SA, Paracha RZ, Ahmad J, Ali A (2015) Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect Genet Evol 32:280–291
Pacheco LG, Slade SE, Seyffert N, Santos AR, Castro TL, Silva WM, Santos AV, Santos SG, Farias LM, Carvalho MA, Pimenta AM, Meyer R, Silva A, Scrivens JH, Oliveira SC, Miyoshi A, Dowson CG, Azevedo VA (2011) Combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol 11(1):12
Pang HY, Li Y, Wu ZH, Jian JC, Lu YS, Cai SH (2010) Immunoproteomic analysis and identification of novel immunogenic proteins from Vibrio harveyi. J Appl Microbiol 109(5):1800–1809
Pellon A, Ramirez-Garcia A, Buldain I, Antoran A, Rementeria A, Hernando FL (2016) Immunoproteomics-based analysis of the immunocompetent serological response to Lomentospora prolificans. J Proteome Res 15(2):595–607
Rappuoli R, Covacci A (2003) Reverse vaccinology and genomics. Science 302:602
Rodríguez-Ortega MJ, Norais N, Bensi G, Liberatori S, Capo S, Mora M, Scarselli M, Doro F, Ferrari G, Garaguso I, Maggi T, Neumann A, Covre A, Telford JL, Grandi G (2006) Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat Biotechnol 24:191–197
Santos AR, Carneiro A, Gala-García A, Pinto A, Barh D, Barbosa E, Aburjaile F, Dorella F, Rocha F, Guimarães L, Zurita-Turk M, Ramos R, Almeida S, Soares S, Pereira U, Abreu VC, Silva A, Miyoshi A, Azevedo V (2012) The Corynebacterium pseudotuberculosis in silico predicted pan-exoproteome. BMC Genom 13(Suppl 5):S6
Seib KL, Zhao X, Rappuoli R (2012) Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin Microbiol Infect 5:109–116
Sengupta N, Alam SI, Kumar B, Kumar RB, Gautam V, Kumar S, Singh L (2010) Comparative proteomic analysis of extracellular proteins of Clostridium perfringens type A and type C strains. Infect Immun 78:3957–3968
Snider CA, Voss BJ, McDonald WH, Cover TL (2016) Growth phase-dependent composition of the Helicobacter pylori exoproteome. J Proteom 130:94–107
Testa JS, Shetty V, Hafner J, Nickens Z, Kamal S, Sinnathamby G, Philip R (2012) MHC class I-presented T cell epitopes identified by immunoproteomics analysis are targets for a cross reactive influenza-specific T cell response. PLoS One 7(11):e48484. doi:10.1371/journal.pone.0048484
Trost M, Wehmhöner D, Kärst U, Dieterich G, Wehland J, Jänsch L (2005) Comparative proteome analysis of secretory proteins from pathogenic and non-pathogenic Listeria species. Proteomics 5:1544–1557
Tsolakos N, Brookes C, Taylor S, Gorringe A, Tang CM, Feavers IM, Wheeler JX (2014) Identification of vaccine antigens using integrated proteomic analyses of surface immunogens from serogroup B Neisseria meningitidis. J Proteom 101:63–76
Vivona S, Bernante F, Filippini F (2006) NERVE: new enhanced reverse vaccinology environment. BMC Biotechnol 6:35
Walters MS, Mobley HL (2009) Identification of uropathogenic Escherichia coli surface proteins by shotgun proteomics. J Microbiol Methods 78(2):131–135
Walters MS, Mobley HL (2010) Bacterial proteomics and identification of potential vaccine targets. Expert Rev Proteom 7:181–184
Walz A, Mujer CV, Connolly JP, Alefantis T, Chafin R, Dake C, Whittington J, Kumar SP, Khan AS, DelVecchio VG (2007) Bacillus anthracis secretome time course under host-simulated conditions and identification of immunogenic proteins. Proteome Sci 5:11
Wen YT, Tsou CC, Kuo HT, Wang JS, Wu JJ, Liao PC (2011) Differential secretomics of Streptococcus pyogenes reveals a novel peroxide regulator (PerR)-regulated extracellular virulence factor mitogen factor 3 (MF3). Mol Cell Proteom 10(M110):007013
Wöhlbrand L, Trautwein K, Rabus R (2013) Proteomic tools for environmental microbiology-a roadmap from sample preparation to protein identification and quantification. Proteomics 13:2700–2730
Wright A, Wait R, Begum S, Crossett B, Nagy J, Brown K, Fairweather N (2005) Proteomic analysis of cell surface proteins from Clostridium difficile. Proteomics 5:2443–2452
Yang HL, Zhu YZ, Qin JH, He P, Jiang XC, Zhao GP, Guo XK (2006) In silico and microarray-based genomic approaches to identifying potential vaccine candidates against Leptospira interrogans. BMC Genom 16(7):293
Yang Y, Wang L, Yin J, Wang X, Cheng S, Lang X, Wang X, Qu H, Sun C, Wang J, Zhang R (2011) Immunoproteomic analysis of Brucella melitensis and identification of a new immunogenic candidate protein for the development of brucellosis subunit vaccine. Mol Immunol 49(1–2):175–184
Zeng L, Zhang Y, Zhu Y, Yin H, Zhuang X, Zhu W, Guo X, Qin J (2013) Extracellular proteome analysis of Leptospira interrogans serovar Lai. OMICS 17(10):527–535
Zhao Z, Yan F, Ji W, Luo D, Liu X, Xing L, Duan Y, Yang P, Shi X, Lu Z, Wang X (2011) Identification of immunoreactive proteins of Brucella melitensis by immunoproteomics. Sci China Life Sci 54(9):880–887
Zielke RA, Wierzbicki IH, Baarda BI, Gafken PR, Soge OO, Holmes KK, Jerse AE, Unemo M, Sikora AE (2016) Proteomics-driven antigen discovery for development of vaccines against gonorrhea. Mol Cell Proteom 15(7):2338–2355
Acknowledgments
We thank Lt. Gen VK Sharma, Vice Chancellor, Amity University, Madhya Pradesh for providing his support
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing or conflicts of interest.
Rights and permissions
About this article

Cite this article
Dwivedi, P., Alam, S.I. & Tomar, R.S. Secretome, surfome and immunome: emerging approaches for the discovery of new vaccine candidates against bacterial infections. World J Microbiol Biotechnol 32, 155 (2016). https://doi.org/10.1007/s11274-016-2107-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-016-2107-3