
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) has continuously mutated since its first isolation in China in 1996, leading to difficulties in infection prevention and control. Infections caused by PRRSV-2 strains are the main epidemic strains in China, as determined by phylogenetic analysis. In this study, we focused on the prevalence and genetic variations of the non-structural protein 4 (NSP4) from PRRSV-2 over the past 20 years in China. The fundamental biological properties of the NSP4 were predicted, and an analysis and comparison of NSP4 homology at the nucleotide and amino acid levels was conducted using 123 PRRSV-2 strains. The predicted molecular weight of the NSP4 protein was determined to be 21.1 kDa, and it was predicted to be a stable hydrophobic protein that lacks a signal peptide. NSP4 from different strains exhibited a high degree of amino acid (85.8–100%) and nucleotide sequence homology (81.0–100%). Multiple amino acid substitutions were identified in NSP4 among 15 representative PRRSV-2 strains. Phylogenetic analysis showed that the lineage 8 and 1 strains, the most prevalent strains in China, were indifferent clades with a long genetic distance. This analysis will help fully elucidate the parameters of the PRRSV NSP4 epidemic in China to lay a foundation for adequate understanding of the function of NSP4. Genetic information results from the accumulation of conserved and non-conserved sequences. The high conservation of the NSP4 gene determines the most basic life traits and functions of PRRSV. Analyzing the spatial structure of NSP4 protein and studying the genetic evolution of NSP4 not only provide the theoretical basis for how NSP4 participates in the regulation of the innate response of the host but also provide a target for genetic manipulation and a reasonable target molecule and structure for new drug molecules.
Similar content being viewed by others


Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) can cause fever and anorexia in infected pigs, late abortion, premature delivery, stillbirth, weak and mummified fetuses in pregnant sows, and death of pigs of all ages (mainly piglets) because of respiratory diseases [1]. Infection with highly pathogenic strains can cause fever, skin cyanosis, dyspnea, and acute death in adult pigs. After PRRSV infects pigs, it causes host immunosuppression, which reduces the level of immunoprotection and immunity of pigs, thereby increasing the possibility of mixed infection involving other pathogens [2]. Consequently, it is difficult to eliminate the infection, which causes substantial losses to China's swine industry.
PRRSV belongs to the order Nidovirale, family Arteriviridae, and is a single-stranded positive-strand RNA virus with an envelope and no segments [3]. RNA synthesis of PRRSV includes the replication of genomic RNA and the synthesis of subgenomic RNA, both of which are mediated by the replication–transcription complex composed of non-structural proteins (NSPs) encoded by the virus (possibly including host factors) [4]. In 1990, PRRSV was successfully isolated in the Netherlands and named the Lelystad Virus strain. The VR2332 strain was isolated in the United States in 1991. There are vast and noticeable differences in the nucleotide sequences of different PRRSV isolates. According to their antigenicity, PRRSV strains are mainly subdivided into PRRSV-1 and PRRSV-2, and most of China's epidemic strains are PRRSV-2 [5]. The nucleotide sequence homology between them is approximately 60%.
In 2010, Shi et al. [6] proposed a systematic PRRSV-2 classification based on the ORF5 gene, dividing PRRSV-2 into 9 lineages and 37 sub-lineages. Based on the global PRRSV classification system, four lineages of the PRRSV-2 strains are prevalent in China. CHsx1401-2014, HNyc15-2015, and NADC30-2008 are lineage 1 representative strains [7]. Lineage 3 is a mutant strain group newly discovered in China in 2010; however, the overall detection rate was not high, and it was common in Fujian, Guangdong, and other areas in southern China, with GM2-2011, QYYZ-2011, and FJFS-2012 as its representative strains [5]. Lineage 5 appeared earlier in China, its representative strain BJ-4 was reported in 1996, and the overall detection rate was not high, which was similar to that of lineage 3. Other representative strains were ATCC VR2332-1992 and RespPRRS MLV-1994. Lineage 8 is similar to lineage 1, which had a high detection rate in China and could be divided into different subgroups, including the classic PRRSV (C-PRRSV) and highly pathogenic (HP-PRRSV) subgroups [8]. The classic type CH-1a-1996 was the first PRRSV strain reported in China, and it was common in China until 2006 [5]. CH-1R-2008 was its attenuated vaccine strain, and CH2002-2009 was its representative strain. Since the outbreak of the HP-PRRSV-like group strain in 2006, this type of strain has been exhibiting an epidemic trend in China, and its pathogenicity is much higher than that of the classical strains. Its representative strains were JXA1-2006, TJ-2006, and HuN4-2006 [9].
The NSPs of PRRSV play essential roles in viral replication and host immune response. During the processing of NSPs precursors (polyproteins pp1a and pp1ab), the PRRSV NSP4 has been proposed to mediate nine cleavages in the NSP3-12 region [10, 11]. Doland [12] reported that the crystal structure of PRRSV 3C-like serine protease (3CLSP) consists of three parts: two anti-parallel β-folding buckets (domain I and II) and a C-terminal α/β domain (domain III). The active region of NSP4 3CLSP is located between domain I and II. A histidine (His) at amino acid position 39, an aspartic acid (Asp) at position 64, and a serine (Ser) at position 118 constitute the His-Asp-Ser catalytic triad, which is an important amino acid site for the 3CLSP activity of NSP4 [13, 14]. Any substitution of these three amino acids will eliminate the protease activity of 3CLSP [15]. The overall primary structure of NSP4 is shown in Fig. A1. The amino acids that form the active site of the enzyme are at positions 39, 64, and 118, and amino acid substitutions at positions 155, 180, and 185 can change the function of the NSP4 protein.
The NSP4 gene is highly conserved, and the NSP4 antibody can be detected in the early stages of PRRSV infection [16]. As an important protease encoded by PRRSV, NSP4 also participates in various immunosuppressive reactions and plays an important role in antagonizing the natural immune response of the host by mechanisms other than antagonization of the type I interferon (IFN-I) signaling pathway. For example, NSP4 can activate the Notch signaling pathway, regulate the antigen presentation pathway, and induce apoptosis and other cell responses to help the virus escape host immunity [17,18,19]. 3CLSP plays an essential role in regulation of PRRSV replication, inhibition of the production of host IFN, and induction of host cell apoptosis. It has, therefore, emerged as an important target protein for further study of the pathogenesis of PRRSV [20,21,22]. NSP4 interacts with many types of host proteins, and it can cleave host proteins, which may be one of the main mechanisms by which PRRSV escapes detection by the host immune response. As one of the main proteases, PRRSV NSP4 plays an important role in virus proliferation, and 3CLSP has become an important target for drug design [23, 24].
In this study, to explore the role of NSP4 in viral immune evasion from the perspective of genetic variation in PRRSV, the structure and possible functions of NSP4 protein were predicted. We analyzed the nucleotides and amino acids of 123 strains of PRRSV-2 NSP4 in general and constructed a phylogenetic tree, and we carefully analyzed the nucleotide and amino acid composition of representative strains of each lineage, to better understand the genetic variation of NSP4, it provides a theoretical basis and reference for the future epidemic trend and prevention and control of PRRS.
Materials and methods
Sequence analysis of PRRSV NSP4
ExPASy (http://web.expasy.org/protparam/) was used to analyze the primary physical and chemical properties of NSP4, and the SignalP 5.1 Server (http://www.cbs.dtu.dk/services/signalp/) was used to predict the signal peptide. Online software SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/secpred_sopma.pl) was used to predict the secondary structure of the NSP4 protein. Online software SWISS-MODEL (http://www.swissmodel.expasy.org) was used to predict the tertiary structure of the NSP4 protein. The HuN4-2006 strain was selected for sequence analysis of PRRSV NSP4, a typical strain with high pathogenicity. The information on the HuN4-2006 strain is shown in Table 1.
Nucleotide sequence alignment of NSP4
The Clustal W method in the MegAlign function of DNASTAR package (version 7.0; DNASTAR, Madison, WI) was used to analyze NSP4 nucleotide sequence similarities, and information regarding the reference strains is provided in Table 1.
Amino acids sequence alignment of NSP4
The Clustal W method in the MegAlign function of DNASTAR software was used to analyze NSP4 amino acid sequence similarities, and information regarding the reference strains is provided in Table 1. The amino acid sequences of representative strains of each lineage were compared using BioEdit Sequence Alignment Editor (version 7.2.6.1).
Phylogenetic tree analysis
The phylogenetic tree was constructed for the genetic evolution analysis of the NSP4 gene using the maximum likelihood (ML) and Bayesian method (BI). The ML method employed MEGA software (version 7.0; Center for Evolution and Informatics, Tempe, AZ). The ML value supports using a general time-reversible model (GTR) with a gamma distribution with invariant sites (G + I) for two databases. The Bayesian method was conducted with PhyloSuite software, and Model Finder automatically configured the optimal model. The Interactive Tree Of Life (https://itol.embl.de) is an online software for the display, manipulation, and annotation of phylogenetic trees [25], and information regarding the reference strains is provided in Table 1.
Results
Sequence analysis
Distribution of bases
The total length of the NSP4 sequence of HuN4-2006 strain was 612 bp, corresponding to 204 amino acids, with 133 adenines (A), accounting for 21.73% of the total number of bases; 175 guanines (G), accounting for 28.59% of the total bases; 151 thymines (T), accounting for 24.67% of the total number of bases; and 153 cytosines (C), accounting for 25.00% of the total number of bases. Thus, the percentage of AT was 46.41%, and the percentage of CG was 53.59%.
Basic physicochemical properties
The DNASTAR software package predicted that the NSP4 gene encodes 16 highly basic amino acids (K and R), 19 highly acidic amino acids (D and E), 73 hydrophobic amino acids (A, I, L, F, W, and V), and 51 polar amino acids (N, C, Q, S, T, and Y).
Stability coefficient and signal peptide prediction
ExPASy analysis showed that the predicted molecular weight of the NSP4 protein of HuN4-2006 strain was 21.1 kDa. The instability coefficient was 20.78, indicating a stable protein. The average hydrophilicity was 0.081, demonstrating that NSP4 was a hydrophobic protein. The SignalP 5.1 server program estimated that the encoded protein did not have a signal peptide.
Secondary structure analysis
An online SOPMA program was used to predict the secondary structure of the PRRSV NSP4 protein. The results show that alpha helix, beta turns, random coils, and extended chain accounted for 11.27%, 10.78%, 45.59%, and 32.35%, respectively (Figure S1C). The SWISS-MODEL was used to construct the possible tertiary structure model of the NSP4 protein of PRRSV. The estimated quality of the global model was 0.92, and the similarity with the template was 99.51%, indicating that the model was reasonably accurate (Figure S1D).
Nucleotide similarity
Comparison and analysis of the NSP4 nucleotides of 123 PRRSV-2 strains showed that the nucleotide homology between NSP4 of 123 PRRSV-2 strains was 81.0%–100%, of which the nucleotide homology between the NADC31-2008 and WUH2-2009 strains was the lowest (81.0%), and some nucleotide sequences were highly similar, with homology as high as 100%. For example, based on information for the 123 PRRSV-2 strains, the TJbd14-1-2014 and 10FS-GD-2010 strains, the GX1003-2010 and SY0608 strains, and the GS-2002-2009 and GS2003-2003 strains have identical nucleotide sequences. The nucleotide sequence of the QY2010-2010 strain was identical to that of the QYYZ-2011 strain, and the sequence of the FZ06-2006 strain was identical to that of the SY0909-2009, JXwn06-2016, JXA1-p120-2009, and HEB1-2006 strains. The nucleotide sequences of the ATCC VR2332-1992 and YN-2011-2011, RespPRRS MLV-1994, SD1-100, PRRSV01-2008, Henan-XIX-2013, BJ-4-1996, and CC-1-2006 strains were identical.
To further explore the genetic variation of NSP4 in PRRSV evolution, the selected representative strains of each lineage were used to analyze nucleotide homology further to understand the nucleotide evolution relationship among the lineages, as shown in Fig. 1.

Nucleotide similarity analysis of NSP4 sequence of 15 representative strains in lineages 1, 3, 5, and 8. The Clustal W method in the MegAlign function of DNASTAR software was used to analyze the similarity of NSP4 nucleotides. The 15 representative strains selected were CHsx1401-2014, HNyc15-2015, NADC30-2008 of lineage 1, GM2-2011, QYYZ-2011, FJFS-2012 of lineage 3, ATCC-VR2332-1992, RespPRRS MLV-1994, BJ-4-1996 of lineage 5, CH-1a-1996, CH-1R-2008, and CH2002-2009 of the C-PRRSV-like group in lineage 8, and JXA1-2006, TJ-2006, and HuN4-2006 of the HP-PRRSV-like group in lineage 8
As shown in Fig. 1, the nucleotide homology in the lineage 1 group was 93.3–96.1%; among the lineage 3 group, it was 99.5–99.7%; among the lineages 5 group, it was 100%; and among the lineage 8 group, it was 90.0–99.8%. The homology of the C-PRRSV-like group was 99.0–99.8%, and that of the HP-PRRSV-like group was 99.7–99.8%.
Amino acid sequence similarity
The amino acids of NSP4 of the 123 PRRSV-2 strains were compared. The results showed that the amino acid homology of each PRRSV-2 ranged from 85.8 to 100%, of which the 15LY02-FJ-2015 strain had lower amino acid homology than that of the SCya18-2018 strain and was the same as that of the FJFS-2012 and WUH2-2009 strains. To further explore the genetic variation of amino acids in NSP4 during PRRSV evolution, representative strains of each lineage were selected to analyze the homology of the amino acid to further elucidate the amino acid evolution relationship among each lineage (Fig. 2).

Amino acid similarity analysis of NSP4 sequence of 15 representative strains in lineages 1, 3, 5, and 8. The Clustal W method in the MegAlign function of DNASTAR software was used to analyze the similarity of NSP4 amino acids. The 15 representative strains selected were CHsx1401-2014, HNyc15-2015, NADC30-2008 of lineage 1, GM2-2011, QYYZ-2011, FJFS-2012 of lineage 3, ATCC-VR2332-1992, RespPRRS MLV-1994, BJ-4-1996 of lineage 5, CH-1a-1996, CH-1R-2008, and CH2002-2009 of the C-PRRSV-like group in lineage 8, and JXA1-2006, TJ-2006, and HuN4-2006 of the HP-PRRSV-like group in lineage 8
As shown in Fig. 2, the amino acid homology among the lineage 1 group was 96.6–97.5%; among the lineage 3 group, it was 99.0–99.5%; among the lineages 5 group, it was 100%; and among the lineage 8 group, it was 94.6–100.0%, among which the homology of the C-PRRSV-like group was 97.5–99.5% and the homology of the HP-PRRSV-like group was 100%.
Amino acid sequence alignment
The amino acids of representative strains of each lineage were compared using the software BioEdit Sequence Alignment Editor (Fig. 3).

Amino acid sequence alignment of NSP4 of 15 representative strains in lineages 1, 3, 5, and 8. Sequence Alignment Editor software was used to analyze the amino acids of NSP4. The 15 representative strains selected were CHsx1401-2014, HNyc15-2015, NADC30-2008 of lineage 1, GM2-2011, QYYZ-2011, FJFS-2012 of lineage 3, ATCC-VR2332-1992, RespPRRS MLV-1994, BJ-4-1996 of lineage 5, CH-1a-1996, CH-1R-2008, and CH2002-2009 of the C-PRRSV-like group in lineage 8, JXA1-2006, TJ-2006, and HuN4-2006 of the HP-PRRSV-like group in lineage 8
Figure 3 shows that the amino acid sequence of NSP4 of PRRSV-2 comprised 204 amino acid residues and that the amino acid sequence of NSP4 is relatively conserved. However, there were amino acid substitutions at several sites. The amino acid sequence between positions 74–86 and 178–187 has a high frequency of amino acid substitution, whereas amino acid substitutions are rare or absent at other positions.
Phylogenetic tree analysis
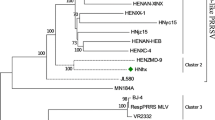
Referring to the reported genotyping study by Shi et al. [6], all the previously identified lineage reference sequences were also clustered in the same lineage in the phylogenetic tree of this study. Phylogenetic tree analysis showed that the lineage 3 strains represented by GM2-2011, QYYZ-2011, and FJFS-2012, and the lineage 1 strains represented by CHsx1401-2014, HNyc15-2015, and NADC30-2008 had a close genetic distance. The strains of lineages 1 and 8, including the classic CH-1a-like and the highly pathogenic HP-PRRSV-like, had a long genetic distance (Figs. 4, 5).

The phylogenetic tree was constructed based on the NSP4 of 123 PRRSV strains. Phylogenetic tree analysis of NSP4 gene constructed using MEGA software (version 7.0; Center for Evolutionary Medicine and Informatics, Tempe, AZ). The maximum likelihood value is supported using the general time-reversible model (GTR) with gamma distribution with invariable sites (G + I) for both databases. The circles (filled circle) indicate the attenuated live vaccine strains or vaccine derivatives. The triangles (filled triangle) refer to the representative virus strains of each lineage in China

The phylogenetic tree was constructed based on the NSP4 of 123 PRRSV strains. Phylogenetic tree analysis of NSP4 gene constructed by PhyloSuite software. After Model Finder automatically configures the optimal model, the phylogenetic tree is constructed by the Bayesian method
Discussion
In 1996, the classic PRRSV CH-1a strain was first isolated in China. From 2008 to 2015, HP-PRRSV was the epidemic strain in China, whereas, after 2015, the NADC30-like strain became the epidemic strain [26, 27]. PRRSV has been continuously transmitted and has undergone mutations for more than 20 years since its discovery in 1996. In the past, amino acid changes in highly variable regions (NSP2 and ORF5) have been the focus of attention. In recent years, various strain recombination events have been identified, with increasing recombination frequencies [28, 29]. Zhao et al. [30] confirmed that JL580 (a NADC30-like strain), HP-PRRSV, and local strains have different mixed genetic backgrounds and are highly pathogenic. Chen et al. [31] experimentally demonstrated that HBap4-2018 is a new PRRSV strain recombined with HP-PRRSV as the major parent strain and NADC30-like PRRSV as the minor parent strain, retaining most of the virulence-related regions in the HP-PRRSV genome, which was highly pathogenic to piglets. Li et al. [32] found that HNyc15 was formed by recombining VR2332 and CH-1a gene fragments. Widespread use of representative attenuated vaccine strains of PRRSV will also increase the diversity of PRRSV in this area [33]. These genetic events indicate that gene recombination plays a critical role in the evolution of PRRSV.
During the evolution of PRRSV, the changes in NSP2 in the highly variable region are apparent. Compared with classical PRRSV, HP-PRRSV (29 + 1 amino acids) and NADC30-like strains have the characteristics of discontinuous deletion (111 + 5 + 1 + 19 amino acids). To fully understand the changes in NSP4 in genetic evolution, the nucleotide and amino acid sequences of NSP4 of the PRRSV reference strain were compared and analyzed using the Clustal W method in the MegAlign function of DNASTAR software. The amino acids of representative strains of each lineage were compared using the software BioEdit Sequence Alignment Editor, which was used for multisequence alignment analysis, and phylogenetic tree analysis of the NSP4 gene was performed using MEGA7.0 software.
NSP4 is a hydrophobic protein and does not have a signal peptide. Its hydrophobicity is conducive to internal protein folding to form α helix secondary structure, which ensures its protein stability. The absence of a signal peptide can indicate that the NSP4 protein is located in the cytoplasm or organelle matrices, but is not a membrane protein or secreted protein. There has been limited basic research to date on NSP4. Analysis of the secondary and tertiary structures of the NSP4 protein and identification of the functional units or domains can provide a target for genetic manipulation and a reasonable target molecule and structure for new drug molecules. The nucleotide and amino acid homology analysis of PRRSV-2 NSP4 showed that the nucleotide homology among 123 PRRSV-2 NSP4 was 81.0–100%, and the amino acid homology was 85.8–100%, thus showing that NSP4 has a high degree of amino acid and nucleotide homology. In Fig. 1, we speculated that the NSP4 sequence has undergone a series of corresponding changes during mutation and recombination of different lineage 1 strains. There is a high degree of nucleotide homology between lineage 3 and 5, and the detection rate for these two lineages has been low in China. We speculated that PRRSV may exhibit genetic variation, with a low probability for evolutionary changes, thus preventing PRRSV from escaping the host immune response. However, the specific reasons for this require further research. Lineages 1 and 8 were the most prevalent strains in China. The lineages 1 and 8 were prone to recombination and adapted to enhance their survival during long-term evolution and mutation. As shown in Fig. 1, the nucleotide homology of the JL580-2013 and the HP-PRRSV-like group strains in the NSP4 sequence was as high as 98%, indicating that the NSP4 sequence may participate in the recombination event and play an important role. The specific events warrant further study. As shown in Fig. 2, the amino acid homology within pedigrees was higher than that between pedigrees, but overall, the amino acid homology of NSP4 was very high. Therefore, we can conclude that although NSP4 has maintained highly conserved characteristics, genetic variations nonetheless still occur in the process of PRRSV continuous evolution. Based on data in Fig. 3 of the comparison and analysis of amino acid sequences, it was concluded that, compared with NSP2 and ORF5, which have extensive genetic variations [34], NSP4 contained no areas of amino acid insertions or deletions, with individual substitutions only occurring at specific sites, and the substitution probability at other sites was very low. Overall, PRRSV NSP4 has been relatively conserved in the process of evolution in China. Previous studies have shown that NSP4 inhibits the production of IFN-β in the host cell's nucleus, and the substitution of NSP4 Thr155 by Lys can enhance the level of NSP4 in the nucleus, which enhances inhibition of IFN-β transcription in vitro [24]. Ser180 of NSP4 is required for degradation of zinc-finger antiviral protein (ZAP) [35]. Thus, substitution of this amino acid at position180 down-regulates the ability of PRRSV to degrade ZAP, thereby antagonizing its antiviral effect. Mutations resulting in substitution of NSP4 Asp185 of HP-PRRSV impair the ability of NSP4 to cleave the essential regulatory protein of the nuclear transcription factor nuclear factor-kappa B and mitochondrial localization antiviral protein (MAVS, also known as IPS-I, VISA, or CARDIF), thereby antagonizing the production of IFN-β [20]. In addition, the sequence of the NSP4 catalytic triad His39-Asp64-Ser118 was highly conserved, and there were no mutations resulting in substitutions at amino acid position 39. There was a very low probability of amino acid substitutions at position 64 but no amino acid substitutions at position 118. Substitution of these amino acids may be related to viral virulence, phagocytosis, and cleavage ability, but this requires further validation. In Figs. 4 and 5, the phylogenetic tree analysis shows that lineages 1 and 8 have a long genetic distance, but lineages 1 and 8 have always been common strains in China, and gene recombination readily occurs between them. Through recombination analysis, Wang et al. [36] found that the HNhx-2016 strain was the result of recombination between NADC30-like and HP-PRRSV-like group strains from NSP4 (nt 5261) to NSP9 (nt 7911). This type of genetic event may also explain why PRRSV constantly mutates and recombines to escape the host immune response, which is conducive to higher survival of PRRSV in the host.
Future perspective
Genetic information is the result of the accumulation of conserved sequences and non-conserved sequences. Conserved sequences determine species, and non-conserved sequences determine intra-species differences. The high conservation of the NSP4 gene determines the most basic life traits and functions of PRRSV, and mutation of the conserved sequence usually leads to very serious consequences. As one of the main proteases, PRRSV NSP4 plays an important role in virus proliferation, and the characteristics of 3CLSP make it a promising target for drug design. Shi et al. [37] screened targeted NSP4 antiviral drugs and obtained two antiviral candidate drugs with remarkable inhibitory activities. Analyzing the spatial structure of NSP4 protein and studying the genetic evolution of NSP4 can help screen key amino acid sites, thereby identifying important antiviral targets for the design of new drug molecules [38], as well as providing a theoretical basis for vaccine development or NSP4 gene detection methods in future.
Whole-genome sequencing has been widely used in pathogen analysis and promotes in-depth study of system dynamics and comprehensive understanding of the diversity of infectious diseases [39]. PRRSV exhibits high mutation and recombination rates. Moreover, the transmission mechanism of PRRSV can be explained from multiple perspectives, such as phylogenetic and evolutionary analysis of specific genes (ORF5,NSP2) and varying rates of nucleotide substitution among different sites [40]. NSP4 plays an important role in antagonizing the innate immune response of the host. Analysis of the genetic variation of NSP4 may provide the theoretical basis for how NSP4 participates in regulation of the innate response of the host. In future, more research should be conducted to elucidate the role of NSP4 in the pathogenesis so as to prevent and control PRRS.
Data availability
All datasets are available in the main manuscript. The dataset supporting the conclusions of this article is included within the article.
References
Sha H, Zhang H, Chen Y, Huang L, Zhao M, Wang N (2022) Research progress on the NSP9 protein of porcine reproductive and respiratory syndrome virus. Front Vet Sci 9:872205. https://doi.org/10.3389/fvets.2022.872205
Lunney JK, Benfield DA, Rowland RR (2010) Porcine reproductive and respiratory syndrome virus: an update on an emerging and re-emerging viral disease of swine. Virus Res 154(1–2):1–6. https://doi.org/10.1016/j.virusres.2010.10.009
Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B et al (2016) Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154. https://doi.org/10.1146/annurev-animal-022114-111025
Song J, Liu Y, Gao P, Hu Y, Chai Y, Zhou S et al (2018) Mapping the nonstructural protein interaction network of porcine reproductive and respiratory syndrome virus. J Virol 92(24):e01112. https://doi.org/10.1128/JVI.01112-18
Guo Z, Chen XX, Li R, Qiao S, Zhang G (2018) The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: a molecular epidemiological perspective. Virol J 15(1):2. https://doi.org/10.1186/s12985-017-0910-6
Shi M, Lam TT, Hon CC, Murtaugh MP, Davies PR, Hui RK et al (2010) Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J Virol 84(17):8700–8711. https://doi.org/10.1128/JVI.02551-09
Paploski IAD, Pamornchainavakul N, Makau DN, Rovira A, Corzo CA, Schroeder DC et al (2021) Phylogenetic structure and sequential dominance of sub-lineages of PRRSV type-2 lineage 1 in the United States. Vaccines (Basel) 9(6):608. https://doi.org/10.3390/vaccines9060608
Li Y, Jiao D, Jing Y, He Y, Han W, Li Z et al (2022) Genetic characterization and pathogenicity of a novel recombinant PRRSV from lineage 1, 8 and 3 in China failed to infect MARC-145 cells. Microb Pathog 165:105469. https://doi.org/10.1016/j.micpath.2022.105469
Han J, Zhou L, Ge X, Guo X, Yang H (2017) Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Vet Microbiol 209:30–47. https://doi.org/10.1016/j.vetmic.2017.02.020
Allende R, Lewis TL, Lu Z, Rock DL, Kutish GF, Ali A et al (1999) North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J Gen Virol 80(Pt 2):307–315. https://doi.org/10.1099/0022-1317-80-2-307
Van Dinten LC, Rensen S, Gorbalenya AE, Snijder EJ (1999) Proteolytic processing of the open reading frame 1b-encoded part of arterivirus replicase is mediated by nsp4 serine protease and Is essential for virus replication. J Virol 73(3):2027–2037. https://doi.org/10.1128/JVI.73.3.2027-2037.1999
Dokland T (2010) The structural biology of PRRSV. Virus Res 154(1–2):86–97. https://doi.org/10.1016/j.virusres.2010.07.029
Xu AT, Zhou YJ, Li GX, Yu H, Yan LP, Tong GZ (2010) Characterization of the biochemical properties and identification of amino acids forming the catalytic center of 3C-like proteinase of porcine reproductive and respiratory syndrome virus. Biotechnol Lett 32(12):1905–1910. https://doi.org/10.1007/s10529-010-0370-1
Ma Z, Wang Y, Zhao H, Xu AT, Wang Y, Tang J et al (2020) Correction: porcine reproductive and respiratory syndrome virus nonstructural protein 4 induces apoptosis dependent on its 3C-like serine protease activity. PLoS ONE 15(2):e0230086. https://doi.org/10.1371/journal.pone.0230086
Ma Z, Wang Y, Zhao H, Xu AT, Wang Y, Tang J et al (2013) Porcine reproductive and respiratory syndrome virus nonstructural protein 4 induces apoptosis dependent on its 3C-like serine protease activity. PLoS ONE 8(7):e69387. https://doi.org/10.1371/journal.pone.0069387
Tian X, Lu G, Gao F, Peng H, Feng Y, Ma G et al (2009) Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV). J Mol Biol 392(4):977–993. https://doi.org/10.1016/j.jmb.2009.07.062
Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J et al (2007) The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol 8(6):592–600. https://doi.org/10.1038/ni1465
Qi P, Liu K, Wei J, Li Y, Li B, Shao D et al (2017) Nonstructural protein 4 of porcine reproductive and respiratory syndrome virus modulates cell surface swine leukocyte antigen class i expression by downregulating beta2-microglobulin transcription. J Virol 91(5):e01755-e1816. https://doi.org/10.1128/JVI.01755-16
Zhang F, Gao P, Ge XN, Zhou L, Guo X, Yang HC (2017) Critical role of cytochrome c1 and its cleavage in porcine reproductive and respiratory syndrome virus nonstructural protein 4-induced cell apoptosis via interaction with nsp4. J Integr Agric 16(11):2573–2585. https://doi.org/10.1016/S2095-3119(17)61670-8
Wei ZY, Liu F, Li Y, Wang HL, Zhang ZD, Chen ZZ et al (2020) Aspartic acid at residue 185 modulates the capacity of HP-PRRSV nsp4 to antagonize IFN-I expression. Virology 546:79–87. https://doi.org/10.1016/j.virol.2020.04.007
Liu Q, Yu YY, Wang HY, Wang JF, He XJ (2020) The IFN-gamma-induced immunoproteasome is suppressed in highly pathogenic porcine reproductive and respiratory syndrome virus-infected alveolar macrophages. Vet Immunol Immunopathol 226:110069. https://doi.org/10.1016/j.vetimm.2020.110069
Aeksiri N, Jantafong T (2017) Structural insights into type I and type II of nsp4 porcine reproductive and respiratory syndrome virus (nsp4 PRRSV) by molecular dynamics simulations. J Mol Graph Model 74:125–134. https://doi.org/10.1016/j.jmgm.2017.03.015
Huang C, Zhang Q, Guo XK, Yu ZB, Xu AT, Tang J et al (2014) Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-kappaB essential modulator. J Virol 88(18):10934–10945. https://doi.org/10.1128/JVI.01396-14
Chen Z, Li M, He Q, Du J, Zhou L, Ge X et al (2014) The amino acid at residue 155 in nonstructural protein 4 of porcine reproductive and respiratory syndrome virus contributes to its inhibitory effect for interferon-beta transcription in vitro. Virus Res 189:226–234. https://doi.org/10.1016/j.virusres.2014.05.027
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49(W1):W293–W296. https://doi.org/10.1093/nar/gkab301
Wang HM, Liu YG, Tang YD, Liu TX, Zheng LL, Wang TY et al (2018) A natural recombinant PRRSV between HP-PRRSV JXA1-like and NADC30-like strains. Transbound Emerg Dis 65(4):1078–1086. https://doi.org/10.1111/tbed.12852
Tian K (2017) NADC30-like porcine reproductive and respiratory syndrome in China. Open Virol J 11:59–65. https://doi.org/10.2174/1874357901711010059
Dong XP, Soong L (2021) Emerging and re-emerging zoonoses are major and global challenges for public health. Zoonoses 1(7):1. https://doi.org/10.15212/zoonoses-2021-0001
Vue D, Qy T (2021) Transmission, origin, pathogenesis, animal model and diagnosis. Zoonoses 1(1):14. https://doi.org/10.15212/zoonoses-2021-0017
Zhao K, Ye C, Chang XB, Jiang CG, Wang SJ, Cai XH et al (2015) Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J Virol 89(20):10712–10716. https://doi.org/10.1128/JVI.01446-15
Chen P, Tan X, Lao M, Wu X, Zhao X, Zhou S et al (2021) The novel PRRSV strain HBap4-2018 with a unique recombinant pattern is highly pathogenic to piglets. Virol Sin 36(6):1611–1625. https://doi.org/10.1007/s12250-021-00453-0
Wang LJ, Guo Z, Qiao S, Chen XX, Zhang G (2016) Complete genome sequence of a mosaic NADC30-like porcine reproductive and respiratory syndrome virus in China. Genome Announc 4(6):e01428. https://doi.org/10.1128/genomeA.01428-16
Cao Y, Ouyang H, Zhang M, Chen F, Yang X, Pang D et al (2014) Analysis of molecular variation in porcine reproductive and respiratory syndrome virus in China between 2007 and 2012. Virol Sin 29(3):183–188. https://doi.org/10.1007/s12250-014-3462-6
Zhou L, Chen S, Zhang J, Zeng J, Guo X, Ge X et al (2009) Molecular variation analysis of porcine reproductive and respiratory syndrome virus in China. Virus Res 145(1):97–105. https://doi.org/10.1016/j.virusres.2009.06.014
Zhao Y, Song Z, Bai J, Liu X, Nauwynck H, Jiang P (2020) Porcine reproductive and respiratory syndrome virus Nsp4 cleaves ZAP to antagonize its antiviral activity. Vet Microbiol 250:108863. https://doi.org/10.1016/j.vetmic.2020.108863
Wang LJ, Wan B, Guo Z, Qiao S, Li R, Xie S et al (2018) Genomic analysis of a recombinant NADC30-like porcine reproductive and respiratory syndrome virus in china. Virus Genes 54(1):86–97. https://doi.org/10.1007/s11262-017-1516-1
Shi Y, Lei Y, Ye G, Sun L, Fang L, Xiao S et al (2018) Identification of two antiviral inhibitors targeting 3C-like serine/3C-like protease of porcine reproductive and respiratory syndrome virus and porcine epidemic diarrhea virus. Vet Microbiol 213:114–122. https://doi.org/10.1016/j.vetmic.2017.11.031
Zhang H, Sha H, Qin L, Wang N, Kong W, Huang L et al (2022) Research progress in porcine reproductive and respiratory syndrome virus-host protein interactions. Animals 12(11):1381. https://doi.org/10.31274/etd-180810-1934
Dudas G, Bedford T (2019) The ability of single genes vs full genomes to resolve time and space in outbreak analysis. BMC Evol Biol 19(1):232. https://doi.org/10.1186/s12862-019-1567-0
Frias-De-Diego A, Jara M, Pecoraro BM, Crisci E (2021) Whole genome or single genes? A phylodynamic and bibliometric analysis of PRRSV. Front Vet Sci 8:658512. https://doi.org/10.3389/fvets.2021.658512
Acknowledgements
We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This research was funded by the National Natural Science Foundation of China (31902279, 31902284), Guangdong Science and Technology Program Project (2021A1515012388, 2017A020208079), and the Guangdong Basic and Applied Basic Research Foundation (2021A1515110322).
Author information
Authors and Affiliations
Contributions
HS and HZ performed the experiments and wrote the manuscript; YZ, QL, QZ, NW, LQ, and HL analyzed the data; LH and MZ conceived and designed the experiments; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests
Ethical approval and consent to participate
All authors read and approved the final manuscript.
Consent for publication
Not applicable
Additional information
Edited by Nicola Decaro.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sha, H., Zhang, H., Luo, Q. et al. Variations in the NSP4 gene of the type 2 porcine reproductive and respiratory syndrome virus isolated in China from 1996 to 2021. Virus Genes 59, 109–120 (2023). https://doi.org/10.1007/s11262-022-01957-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-022-01957-x