
Abstract
Avian influenza virus (H9N2) infection is a major problem of product performance in poultry worldwide. Vaccination is used to limit spread, but more knowledge is needed on the epidemiology of virus subtypes to improve vaccine design. In this study, 40 H9N2 subtype avian influenza viruses (AIVs) were isolated from vaccinated poultry flocks in China from 2010 to 2011. Hemagglutinin (HA) from different virus strains was sequenced and analyzed. We found that the HA genes of these strains shared nucleotide and deduced amino acid homologies that ranged from 90.1 to 92.9 and 91.4 to 95.0 %, respectively, when compared with vaccine strains. Phylogenetic analysis showed that the strains tested could be divided into two major groups. Group I consisted of 24 strains isolated mainly from Eastern and Central China. Group II consisted of 20 strains isolated from Southern China. The cleavage site within the HA protein contained two basic motifs, PSRSSR↓GLF for group I, and PARSSR↓GLF for group II. Additional potential glycosylation sites were found at amino acid position 295 in the HA1 of the isolates in group I, compared with isolates in group II and the vaccine strains. Furthermore, 38 out of the 40 isolates had a leucine residue at position 216 (aa 226 in H3), which was characteristic of human influenza virus-like receptor specificity. In the present study we found that geographical factors play a significant role in virus evolution, and emphasize the importance of continuing surveillance of H9N2 AIVs in chickens in China.
Similar content being viewed by others
Introduction
Millions of cases of avian influenza virus (AIV) infection are reported each year. These infections can result in thousands of incidences of death in the population and considerable economic loss to communities worldwide [1, 2]. Influenza viruses are classified into different subtypes partly based on their type of hemagglutinin (HA). Highly pathogenic influenza viruses (HPAIV) are confined to the H5 and H7 virus subtypes that cause severe disease with high mortality, whereas the H9N2 subtype AIVs are referred to as low-pathogenic influenza viruses (LPAIV) and usually cause only mild to moderate disease. Serious outbreaks caused by H9N2 subtype AIVs, however, have been reported to cause severe morbidity and mortality in poultry as a result of co-infection with other pathogens [3, 4].
The H9N2 subtype AIVs are widespread and isolates have been reported worldwide, with the first infectious case in turkeys reported in Wisconsin in 1966 [5]. In mainland China, H9N2 AIV was first isolated from chickens in Guangdong Province in 1994 [6]. Subsequently, the virus has spread rapidly to several other provinces in China [7]. Previous studies have defined two distinct lineages of H9N2 influenza viruses, which include the North American type and the Eurasian type. The Eurasian lineage can be further divided into three major sublineages: the G1 lineage, represented by A/quail/HongKong/G1/97 (HK-G1); the Y280 lineage, represented by A/duck/Hong Kong/Y280/97 (HK-Y280); and the Y439 lineage, represented A/duck/HongKong/Y439/97 (HK-Y439) [8, 9].
Hemagglutinin is the major envelope glycoprotein present in influenza A viruses and is the predominant inducer of neutralizing antibodies during both natural infection and following vaccination [10, 11]. To date, vaccination has been used to limit serious outbreaks of H9N2 subtype AIVs. At present, current vaccines that are based on inactivated viruses elicit a potent immune response against viruses that are closely matched to the vaccine strains [12]. In China, inactivated vaccines derived from earlier strains of virus have been widely used in chicken farms to limit disease [13]. However, serious disease outbreaks have continued to occur in vaccinated flocks [14]. Moreover, some avian H9N2 viruses harbor receptor-binding characteristic present in human strains of the virus, a factor that has increased the potential for virus reassortment in both human and pig respiratory tracts and therefore the potential of transmission to other species [15]. The aim of this study was to determine the phylogenetic relationships and the molecular characterizations of HA genes of H9N2 subtype AIVs that circulated in China from 2010 to 2011 to increase our knowledge on the prevalence of the virus, and to provide useful guidelines for the epidemiological control of H9N2 subtype avian influenza in China.
Materials and methods
Viruses
Forty H9N2 influenza virus strains were isolated from chickens in vaccinated poultry flocks in Hainan, Zhejiang, Guangdong, Guangxi, Fujian, Jiangsu, Guizhou, Hubei, and Anhui provinces in China in the period between April 2010 and October 2011. The chickens were 15–70 days old when examined, and showed typical respiratory symptoms, with a 1–35 % mortality in the isolated cases. The virus strains used in this study were propagated in specific pathogen-free embryonated chicken eggs (ECE). Hemagglutination assays and neuraminidase inhibition tests were performed to subtype the virus, and antisera that were specific for the influenza virus reference strains were used [16]. Allantoic fluid that contained infectious virus was harvested from ECEs and used as a source of stock virus for further analysis.
Viral RNA extraction and one-step RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, USA); total RNA extraction was performed as described in the manufacture’s instruction. Reverse transcription polymerase chain reaction (RT-PCR) was carried out using the PrimeScript®One-Step RT-PCR Kit (TaKaRa Biotechnology, Dalian, China) in 25-μL reaction volumes that contained 20 μL of RT-PCR PreMix (reaction buffer, dNTPs, 2 μL of enzyme mix), 2 μL of extracted viral RNA and 1.5 μL (10 pmol) each of primers specific for HA gene (H9-F: 5′-CAAGATGGAAGTAGTATCACT-3′, and H9-R: 5′-TTGCCAATTATATACAAATGT-3′). The RT-PCR program cycling conditions were 50 °C for 30 min, 94 °C for 2 min, 30 cycles of 94 °C for 40 s, 53 °C for 40 s, 72 °C for 2 min, respectively, followed by an extension step of 72 °C for 10 min.
Gene sequence and phylogenetic analyses
PCR products were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Inc., USA) in accordance with manufacture’s recommendations and the product was cloned into the pMD-19T vector and sequenced (TaKaRa Biotechnology, Dalian, China). The nucleotide sequences were used for comparison with representative H9 strains (Table 1) using the ClustalX 2.0 program. A phylogenetic tree was constructed using MEGA4.1 software with the neighbor-joining method [17], bootstrap values were calculated on 1,000 replicates for alignment.
Nucleotide sequence accession numbers
The 40 HA nucleotide sequences tested in this study were deposited in GenBank under accession numbers JF714998–JF715005, JF715010–JF715016, JF715024–JF715036, JN986883–JN986894.
Results
Homology analysis
The open-reading frames (ORF) of the 40 HA genes were sequenced and contained 1683 nucleotides that coded for 560 amino acids. The nucleotide and deduced amino acid sequence similarities among the tested strains ranged from 88.2 to 100 and 92.3 to 100 %, respectively. The identity of the nucleotide and deduced amino acid sequence among the 40 isolates were 90.1–92.9 and 91.4–95.0 %, respectively, compared with the vaccine strains of H9N2 AIV (A/chicken/Guangdong/SS/94, A/chicken/Shandong/6/96, and A/chicken/Shanghai/F/98). Our results revealed that most of tested strains had non-negligible genetic distances compared with the vaccine strains. The tested strains shared higher nucleotide and deduced amino acid homologies that ranged from 92.1 to 93.5 and 93.0 to 95.4 %, respectively, with the representative strain HK-Y280, which indicated that they belonged to the Y280-like lineage phylogenetically.
Phylogenetic analysis
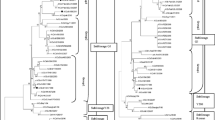
The HA genes of the 40 isolates and 16 reference H9N2 AIV strains were used for phylogenetic analysis. As shown in Fig. 1, all tested strains and vaccine strains belonged to sublineage h9.4.2 as determined by a new nomenclature system [18], which covered A/chicken/Beijing/1/94-like strains and HK-Y280-like strains.

Phylogenetic tree of H9N2 avian influenza viruses (AIVs) isolated in China from 2010 to 2011 based on the viral hemagglutinin sequences. The lineages and sublineages of the viruses were designated based on a new nomenclature system [18] with some representative H9N2 AIVs isolated in other regions of the world. The tested strains in this study were marked with triangles, vaccine strains in mainland China were marked with squares, and the isolates marked with stars stand for representative strains of sublineage grouping
Based on the phylogenetic relationship, the 40 isolates could be divided into two groups. Group I consisted of 24 strains, the majority of which (20/24) were isolated from Eastern China (Anhui, Jiangsu, Zhejiang, and Fujian province, 16 strains) and from Central China (Hubei province, four strains). Group II consisted of 20 strains that were isolated from Southern China (Guangdong, Guangxi, and Guizhou province). These results indicated that the H9N2 subtype AIVs that were isolated in different geographical regions also displayed complexity and diversity.
Mutation analysis
The amino acid sequence of the cleavage site of H9N2 isolates in China has been described in detail previously [14]. In brief, the isolates were distributed into the same two groups, based on the cleavage site amino acid motif (Table 2) and by phylogenetic association. Group I, which exhibited cleavage site motif PSRSSR↓GLF, was comprised of viruses isolated from Eastern and Central China. The group II cleavage site motif was PARSSR↓GLF, except in the case of one Guangdong isolate, A/chicken/Guangdong/FZH/2011, which had the cleavage site motif PAKSSR↓GLF. The vaccine strains (A/chicken/Shandong/6/96, A/chicken/shanghai/F/98) and representative strains (HK-Y280, HK-G1) possessed the same cleavage site motifs as group II. All isolates lacked multiple basic amino acids at the cleavage sites, which met the characteristic of low-pathogenic avian influenza.
Potential glycosylation sites (PGS) that possessed an N–X–T/S motif (where X can be any amino acid except proline) on the mature peptide of HA of the tested strains were analyzed. The results showed that the strains in group I carried eight PGS that were located at amino acid positions 11, 123, 200, 280, 287, and 295 in the HA1 molecule, and at 154 and 213 amino acids in the HA2 molecule (Table 3). However, the isolates in group II did not have a PGS at position 295 of HA1, and carried only seven cleavage sites, which was similar in number to the vaccine strains (A/chicken/Guangdong/SS/94, and A/chicken/Shanghai/F/98) and the representative strain (HK-Y280).
Amino acids that are associated with the receptor-binding activity of the HA proteins constitute the receptor-binding pocket [12]. Most of these amino acid residues were conserved among the test strains (Table 2). Two residues at position 180 from receptor-binding site and position 216 from left-edge of binding pocket displayed remarkable variation: A180 V (or T) and L216Q, respectively as compared with the vaccine strains. In total, 38 isolates had a leucine residue at position 216 (amino acid 226 in H3), which was a characteristic of human influenza virus-like receptor specificity, instead of a glutamine at position 216 as in the vaccine strains.
Discussion
The described characterization of the H9N2 subtype AIVs was based on 40 viruses that were isolated in China from April 2010 to October 2011. In total, 18 out of the 40 viruses were isolated in winter, and 14 strains were isolated in spring. However, only one strain was isolated in summer. These epidemic strain characteristics suggested that H9N2 subtype AIVs usually spread in winter and spring when it is cold and wet.
Vaccination is an effective method to prevent AIVs epidemic, but the vaccine should be effective against current local strains [19, 20]. However, the prediction of which strains will dominate annually is difficult, and mismatches between the vaccine and circulating viruses lead to little or even no protective effect [21]. The H9N2 vaccine strains used in mainland China were selected from viruses isolated in 1990s, after the period of initial outbreak. In this study, the nucleotide sequence analysis of the HA gene showed that the tested strains shared only 90.1–92.9 % homology when compared with the vaccine strains. Previous studies have described that most H9N2 subtype AIVs isolated in the past decade in China belonged to sublineage h9.4.2 [22]. Nevertheless, based on the phylogenetic analysis of HA genes, the tested strains were not homogeneous and could be divided into two subgroups, either associated with the geographical regions of their isolation, and/or on the type of amino acid motifs at their cleavage sites. Phylogenetic analysis showed that the tested strains were derived from HK-Y280, and had a distant genetic relationship with the vaccine strains. All our findings suggested that the vaccine strains need to be update and the candidate virus strains should be selected from the presumably predominant clade.
N-linked glycosidic motifs have been found to play a vital role in mediation of virus infectivity and cell-associated host immune responses [23]. In general, H9N2 subtype AIVs vary in the number of glycosylation sites on HA (six in duck viruses and as many as eight in quail and chicken viruses), no single specific glycosylation site has been found to correspond to adaptation to land-based poultry [24]. In this study, an additional PGS appeared at amino acid position 295 in the HA1 of the isolates in group I, compared with the isolates in group II and the vaccine strains, and may enhance pathogenicity.
Amino acid differences in the receptor-binding site of HAs have been shown to be associated with differences in receptor-binding specificity [25]. It was found that the AIVs had histidine, glutamate, and glutamine at amino acid positions 173, 180, and 216 (amino acids 183,190, and 226 in H3), respectively, at the receptor-binding site [26]. All tested strains had H173, but none of the isolates was had a glutamate residue at amino acid position 180. Twenty-eight out of the 40 isolates had A180, as found in the vaccine strains, eight of the viruses had V180, and the other four had a threonine residue at this set position. These results were in accordance with findings for other H9N2 viruses isolated in China [27]. The influenza viruses with HA proteins that have L216 (amino acid 226 in H3) preferentially bind to the NeuAca2, 6-Gal linkage, whereas those that have Q216 bind to the NeuAca2,3-Gal linkage [28, 29]. A change from L216, typical of the HAs of early human H3N2 viruses, to Q216, typical of HAs of many avian viruses, can cause a switch in host specificity [25]. It had been reported that the proportion of H9N2 subtype AIVs isolates with L216 was 34.3 % in the 1990s and 76 % in the 2000s in China [22, 30]. For the Israeli H9N2 subtype AIVs, L216 occurred in 38 % of the viruses isolated from turkeys and 83 % of the viruses isolated from chickens during the 2000–2005 epizootic [31]. Our study highlights that 95 % (38/40) of the chicken virus isolates had an HA protein with a L216 residue, as these viruses might have the potential to cross species to infect humans, continuous monitoring is required.
In the present study, our findings demonstrated the continued circulation of H9N2 AIVs in chickens in China and showed that different geographical factors played a significant role in virus evolution. Furthermore, the genetic disparity between the prevalent strains and the current vaccine strains should be taken into account when a prevention and control policy is established. Therefore, this study manifested the importance of reinforcement of influenza surveillance in chickens to understand the emergence of potentially pandemic strains in China.
References
D.J. Alexander, Avian Dis. 51, 161–166 (2007)
I.H. Brown, Avian Dis. 54, 187–193 (2010)
I.H. Brown, J. Banks, R.J. Manvell, S.C. Essen, W. Shell, Dev. Biol. (Basel) 124, 45–50 (2006)
H. Nili, K. Asasi, Avian Pathol. 31, 247–252 (2002)
P.J. Homme, B.C. Easterday, Avian Dis. 14, 66–74 (1970)
B.L. Chen, Z.J. Zhang, W.B. Chen, Chin. J. Vet. Med. (Chinese) 22, 3–5 (1994)
K.S. Li, K.M. Xu, J.S. Peiris, L.L. Poon, K.Z. Yu, J. Virol. 77, 6988–6994 (2003)
Y. Guan, K.F. Shortridge, S. Krauss, R.G. Webster, Proc. Natl. Acad. Sci. USA 96, 9363–9367 (1999)
M.N. Matrosovich, S. Krauss, R.G. Webster, Virology 281, 156–162 (2001)
B.E. Johansson, D.J. Bucher, E.D. Kilbourne, J. Virol. 63, 1239–1246 (1989)
J.J. Skehel, D.C. Wiley, Annu. Rev. Biochem. 69, 531–569 (2000)
D.C. Ekiert, R.H. Friesen, G. Bhabha, T. Kwaks, M. Jongeneelen, Science 333, 843–850 (2011)
Z.Q. Wu, J. Ji, K.J. Zuo, Q.M. Xie, H.M. Li, Poult. Sci. 89, 1136–1143 (2010)
C. Li, K. Yu, G. Tian, D. Yu, L. Liu, Virology 340, 70–83 (2005)
T. Saito, W. Lim, T. Suzuki, Y. Suzuki, H. Kida, Vaccine 20, 125–133 (2001)
A.P. Kendal, J.J. Skehel, Concepts and Procedures for Laboratory-Based Influenza Surveillance (Centers for Disease Control and Prevention and Pan-American Health Organization, Atlanta, 1982), pp. B17–B35
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24, 1596–1599 (2007)
S. Liu, K. Ji, J. Chen, D. Tai, W. Jiang, PLoS ONE 4, e5022 (2009)
S. Sambhara, G.A. Poland, Lancet 367, 1636–1638 (2006)
E. Montomoli, I. Manini, Vaccine 25, 1921–1922 (2007)
S. Salzberg, Nature 454, 160–161 (2008)
K. Ji, W.M. Jiang, S. Liu, J.M. Chen, J. Chen, J. Virol. Methods 163, 186–189 (2010)
J.N. Arnold, M.R. Wormald, R.B. Sim, P.M. Rudd, R.A. Dwek, Annu. Rev. Immunol. 25, 21–50 (2007)
D.R. Perez, W. Lim, J.P. Seiler, G. Yi, M. Peiris, J. Virol. 77, 3148–3156 (2003)
Y. Suzuki, Biol. Pharm. Bull. 28, 399–408 (2005)
E. Nobusawa, T. Aoyama, H. Kato, Y. Suzuki, Y. Tateno, Virology 182, 475–485 (1991)
J.H. Liu, K. Okazaki, A. Mweene, W.M. Shi, Q.M. Wu, Virus Genes 29, 329–334 (2004)
G.N. Rogers, J.C. Paulson, R.S. Daniels, J.J. Skehel, I.A. Wilson, Nature 304, 76–78 (1983)
W. Weis, J.H. Brown, S. Cusack, J.C. Paulson, J.J. Skehel, Nature 333, 426–431 (1988)
Y.P. Lin, M. Shaw, V. Gregory, K. Cameron, W. Lim, Proc. Natl Acad. Sci. USA 97, 9654–9658 (2000)
S. Perk, N. Golender, C. Banet-Noach, E. Shihmanter, S. Pokamunsky, M. Pirak, Y. Tendler, M. Lipkind, A. Panshin, Comp. Immunol. Microbiol. Infect. Dis. 32, 221–238 (2009)
Acknowledgments
This research was supported by a grant from the Science and Technology Planning Project of Guangdong Province China (No. 2010B090301019).
Author information
Authors and Affiliations
Corresponding author
Additional information
Feng Chen and Zhuan-Qiang Yan contributed equally to this study.
Rights and permissions
About this article
Cite this article
Chen, F., Yan, ZQ., Liu, J. et al. Phylogenetic analysis of hemagglutinin genes of 40 H9N2 subtype avian influenza viruses isolated from poultry in China from 2010 to 2011. Virus Genes 45, 69–75 (2012). https://doi.org/10.1007/s11262-012-0742-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0742-9