
Abstract
A Chinese isolate of avian infectious bronchitis virus (IBV) designated HH06 was isolated from the kidney tissues of a chicken flock experiencing an outbreak of nephritis. In vivo pathogenicity of the IBV isolate HH06 was determined by inoculating specific pathogen-free (SPF) chickens. The clinical signs and related gross lesions of HH06 infected chickens were similar with those of the field-infected chickens. SPF embryonated eggs were inoculated with virus suspension for serial passage and their genomic RNA was extracted. RT-PCR technique was utilized to amplify the M gene sequence encoding membrane protein of IBV. Recombinant plasmid named T-vector-M was constructed via inserting the M gene into the TA cloning vector, pMD 18-T. The sequenced M gene and its deduced amino acid (aa) sequences were compared with the published sequences of reference strains. The M gene is of 687 bp in length encoding the M protein of 228 amino acids with a predicted molecular weight of 25.4 kDa. The sequences of the M gene and M protein share 83.9–97.9% and 83.6–96.5% homologous identities, respectively, compared with 29 IBV reference strains derived from different regions or countries, which revealed that there are still significant variations between strains. Furthermore, a phylogenetic tree based on these M DNA sequences was generated, and the tree topology suggests that some Chinese IBV strains may have a common ancestor; however, HH06 is a new local IBV isolate that is responsible for the field outbreak of nephritis.
Similar content being viewed by others

Introduction
Avian infectious bronchitis virus (IBV) is the causative agent of infectious bronchitis (IB) and leads to a great economic loss to the poultry industry worldwide. IB has been reported frequently in China since it first appeared in 1965 [1], although some vaccines have been adopted. The variation of serotypes of prevalent IBV may decrease the effect of some IBV vaccines used in some countries. Currently, the Chinese IBV isolates H120 and H52 are commonly used vaccine strains in China. The application of these vaccines in most of the chicken farms has decreased the large-scale outbreak of IB in China, however, the sporadic occurrences of IB are reported frequently. The primary target of IBV is the respiratory tract, and some isolates also replicate in the kidney and oviduct, resulting in nephritis and reduced egg production, respectively [2].
IBV is a member of the family Coronaviridae, which consists of some enveloped viruses that replicate in the cell cytoplasm and contain an unsegmented, single-stranded, positive-sense RNA genome of about 27–32 kb [3–8]. IBV has three major structural proteins, which include the spike (S), the membrane (M), and the nucleocapsid (N) protein. The structural genes, together with other open reading frames (ORFs), organize the typical avian coronavirus genome characterized in order of 5′-Pol-S-3a-3b-E-M-5a-5b-N-UTR-3′ [9]. The spike of IBV is formed by post-translational cleavage of two separate polypeptide components, designated S1 and S2 [10]. The S1 glycoprotein is associated with virus attachment and is a major target of the neutralizing antibodies in chickens. It is commonly accepted that the serotype of IBV is determined by the S gene, particularly the hypervariable S1 gene. The N protein of IBV is the preferred protein to use in development of group-specific serologic assays and is produced abundantly during infection, and has high immunogenicity, readily inducing antibodies and cytotoxic T-lymphocyte immunity in chickens [11].
The M protein, a polytopic protein, is the most abundant component of coronavirions [12]. When synthesized in the absence of other viral components, M protein tends to accumulate in the Golgi complex as detergent-insoluble, polymeric structures, presumably as part of its retention mechanism [13, 14]. The M protein as the major inducing component has been proposed to play a role in interferon induction [15, 16]. It was demonstrated that the coexpression of M and E proteins allowed the formation of pseudoparticles, which exhibited an interferogenic activity similar to that of complete virions [17].
Recently, we isolated an IBV strain (HH06) from northeastern China and cloned its S1 gene, and found this virus has over 98.8% S1 identity with 19 other Chinese IBV isolates. However, the IBV S1 region displays very distinctive difference with the vaccine strain H120 that is used for vaccination in northeastern China. There are several papers describing the epidemic situation regarding IBV in China, based on the S1 gene sequences [18–21]. Evidently, the genotypic and phenotypic characterization of IBV is important for analyzing IBV variations [22]. In this paper, we further characterized the IBV isolate and cloned the M gene. The phylogenetic relationships among the IBV isolates derived from different countries or regions were investigated by sequence comparison and phylogenetic tree topology. This study provides useful information of a Chinese isolate of IBV, which may assist in elucidating the genetic evolution of IBV in China.
Materials and methods
Virus isolation and in vivo pathogenicity
An avian IBV was isolated from the suburb of Harbin, capital of Heilongjiang Province, P.R. China. The IBV designated HH06 was isolated from the kidney tissues of suspected young chicken flock experiencing an outbreak of nephritis, depression, and slight respiratory signs. The virus suspension was prepared from homogenized kidney tissue after freezing and thawing three times. Nine-day-old SPF embryonated eggs were inoculated with the virus suspension (0.3 ml/embryo) containing 100 U penicillin and 100 mg streptomycin/ml via the allantoic cavity and then were incubated at 37°C for 72 h. The harvested allantoic fluid was cleared by centrifugation at 3,000 rpm at 4°C for 15 min and used as inoculum for serial passage (0.1 ml/embryo) up to eight times. Egg lethal dose (ELD)50 of the 8th passage viruses was detected using the embryonated eggs.
For in vivo pathogenicity analysis, 20-day-old SPF chickens were randomly divided into two groups with 15 birds in the experimental group and 5 birds in the control group. Each bird in the experimental group was orally inoculated with 0.5 ml of the allantoic fluid containing the isolate HH06 (105.7 ELD50/ml), and the birds in the control group were orally inoculated with 0.5 ml sterilized physiological saline. All chickens were reared in the isolaters. The infected birds were observed for clinical signs and disease course, and were later killed by intravenous inoculation of barbiturate for observation of gross lesions and sampling of kidney, trachea, and lung.
Extraction of viral RNA and primers
Viral RNA was extracted using TRIzol reagent (TaKaRa, Japan) according to the manufacturer’s instructions. A pair of primers was designed using Primer Premier 5.0 software, and synthesized by TaKaRa Biotechnology Company (Dalian, China) based on the sequence of Beaudette strain of IBV (GenBank accession No. AJ311362). The sense primer, pM1: 5′-TAACGAGTTTCCTAAGAACGGTTGG-3′, and the anti-sense primer, pM2: 5′-ACATTTATGTGTAAAGACTACTTCC-3′, flanking the M gene were used to amplify a 735 bp fragment.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RT-PCR was performed using an RT-PCR Kit (TaKaRa, Japan) according to the manufacturer’s instructions. Each reverse transcription volume had a total volume of 10 μl and used 1 μl total RNA (1 μg/μl) as template. The cDNA obtained was amplified with primers pM1 and pM2. PCR was in 50 μl volumes containing 5 μl of 10× buffer, 3 μl dNTP mixture (2.5 mM), 10 μl cDNA, 1 μl pM1, 1 μl pM2, lμl ExTaq polymerase (TaKaRa, Japan), and 29 μl sterile water. The cycling parameters for the PCR included 94°C for 1 min, 30 cycles at 94°C for 1 min, 49.9°C for 1 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. PCR products were run in 1% agarose gel electrophoresis containing ethidium bromide (0.5 μg/ml) and visualized by subsequent UV transillumination.
Cloning and DNA sequencing
PCR products of each RT-PCR were purified using a Gel Extraction Mini Kit (Omega, USA). Purified PCR products ligated with a TA cloning vector, pMD18-T (TaKaRa, Japan) were transformed into competent cells (strain JM109). Cells carrying recombinant plasmid were screened on Luria-bertani (LB) agar plates containing Ampicillin (50 μg/ml). Colony PCR was utilized to select recombinant bacteria. The PCR conditions were the same as that for the above-mentioned PCR amplification. Three positive clones were randomly selected and cultured. Plasmid DNA prepared by Plasmid Extraction Kit (Omega, USA) was verified with restriction enzyme digestion. The resulting plasmids were designated T-vector-M and sequenced by Sangon bio-company (Shanghai, China).
Sequence comparison and phylogenetic tree analysis
Sequences of T-vector-M and their deduced amino acids were compared with that of 29 IBV reference strains derived from different regions or countries using the Lasergene software package V5.0 (DNASTAR Inc, USA). The composition of the nucleotide sequence of M gene was analyzed with DNAMAN software version 4.0 (Lynnon BioSoft, Quebec, Canada). The IBV origin places and the GenBank accession numbers of these sequences are summarized in Table 1. A phylogenetic tree was generated using an alignment of gene sequences in the M region from the above-mentioned IBV strains by the Lasergene software package V5.0 (DNASTAR Inc, USA).
Results
Isolation and in vivo pathogenicity
Ten-day-old SPF embryonated eggs were inoculated with virus suspension for serial passage. The gross lesions of the infected embryos were hemorrhage on the legs, mottled necrosis of the liver, swelling of the kidneys, dwarfing, and curling. In the eighth passage, egg lethal dose (ELD)50 of the virus isolate was 105.7 ELD50/ml (Table 2).
Twenty-day-old chickens inoculated with the 8th passage viruses exhibited clinical symptoms including depress, cough, sneeze, dyspnea, and water-like feces 5 days post-inoculation. Two chickens died 7 days post-inoculation (PI) and another two chickens died 9 days PI. Then more chickens died continuously, with a mortality rate of 60%. Necropsy showed that the kidney of the dead birds was swollen and exhibited severe urate deposition. In addition, there were large volumes of mucilage in the bronchi of the dead chickens, and their throats appeared slightly hyperemic. The control chickens did not show any clinical symptoms.
Cloning of IBV M gene
The integrity of the RNA extracted was confirmed by denaturing agarose gel electrophoresis, and the total RNA concentration was 1 μg/μl approximately. Using the specific primers for the M gene of IBV, RT-PCR products of approximate 750 bp were successfully amplified (data not shown). The amplicons were cloned into pMD18-T vector, and three positive clones identified by colony PCR were subjected to plasmid extraction and DNA sequencing.
Sequence analysis and comparison
Sequencing results showed that the nucleotide sequences of the three M genes of IBV strain HH06 had no mutation. The consensus sequence was reported to GenBank Database and was assigned an accession number, EF397564. Sequence analysis indicated that the nucleotide sequence of the entire M gene of IBV strain HH06 was 687 pair of bases in length and had a base composition of 196 adenine (29%), 123 cytosine (18%), 151 guanine (22%), and 217 thymine (32%), and a GC content of 40%. Moreover, there were several TATA motifs (possible introns) in the M gene sequence. The M gene sequence is accessible through the website link: http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=125664056. The nucleotide and deduced amino acid sequences of the M gene of strain HH06 were compared with the reference strains derived from different regions or countries. The homology of the HH06 M gene shared 83.9–97.9% and 83.6%–96.5% sequence identity with the 29 IBV reference isolates at the nucleotide level and at the amino acid level, respectively (Table 3). The amino acid sequence alignment showed that various sequences of IBV isolates were of unequal length (data not shown). Although the nucleotide sequence of the HH06 M gene was similar to those of other IBV strains, there were still significant variations between strains in terms of the homologous identity. The Chinese isolate HH06 showed very high sequence homology to many other Chinese isolates, although they were isolated from the different geographic places in China. For example, it has very high sequence homology (more than 96% identity at the nucleotide level) with isolates SAIBK (Sichuan, China), LDT3 (Guangdong, China), CK/CH/LSC/99I (Sichuan, China), and partridge/GD/S14/2003 (Guangdong, China). Interestingly, HH06 had a lower sequence homology with H120 (89.2% at the nucleotide level), although HH06 was isolated from H120 vaccinated chicken.
Phylogenetic tree analysis
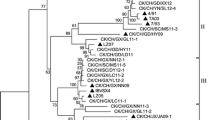
To understand the phylogeny of IBV strains, a phylogenetic tree based on the nucleotide sequences of the M genes was generated by the MEGALIGN program in DNAStar with the Jotun Hein method [23]. Most Chinese IBV isolates showed closer relationships than the foreign isolates according to the branch distribution of the phylogenetic tree, although several strains isolated in the same country, such as, isolates HK and Jilin were clustered into different positions of the phylogenetic tree. However, many Chinese IBV isolates showed closer relationships with the HH06, a northern isolate of China. In contrast, many Chinese isolates did not show a closer relationship with H120, H52, or Mass41 that are widely used vaccine strains in China (Fig. 1).

Phylogenetic relationships between the IBV isolates are constructed on the nucleotide sequences of the M gene using the MEGALIGN program in DNAStar with the Jotun Hein method (Higgins and Sharp 1988 [23]). Symbol “-” used in the isolate name is identical to the “/” in the isolates. The origin places of these IBV isolates are indicated in black bold
Discussion
The SPF chickens inoculated with isolate HH06 exhibited very similar clinical signs and disease course with those from field infected birds, giving the evidence that HH06 was a local epidemic IBV isolate. In addition, the SPF embryonated eggs infected with HH06 virus showed the gross lesions such as hemorrhage on the legs, mottled necrosis of the liver, swelling of the kidneys, and dwarf which were similar with those of the infected with nephropathogenic IBVs [24, 25]. In our previous report, we cloned and analyzed the S1 gene of the isolate HH06 [26]. The HH06 isolate showed high sequence identity with some of the published IBV isolates, particularly some nephropathogenic IBVs. The current study indicated that HH06 was the causative agent of chicken nephritis that was responsible for the field outbreak recently.
Nephropathogenic IBV usually damages kidneys and reproductive tract of chicken, causing a high mortality rate in young flocks. Currently the major control measures of IB are vaccination of live attenuated IBV vaccine (Mass serotypes) such as H52, H120, and Ma5 strains in China; however, the chicken flocks vaccinated with the live vaccines usually fail to present full protection to field virulent IBV challenge, and the vaccinated chicken flocks were often subjected to the attack of nephropathogenic IBV. [25]. In our study, the nephropathogenic IBV isolate HH06 was also isolated from a chicken flock vaccinated with live attenuated vaccine (H120 strain) and experiencing IBV infection, supporting the notion that Mass serotype of live attenuated IBV vaccine might be not ideal in protection of chickens in some places in China.
Genotyping on the basis of the S1 gene sequence is the most common way to classify IBV isolates. There have been several papers describing the epidemic situation regarding IBV in China [19, 21]. We have sequenced the S1 gene of the HH06 strain. The sequencing results indicated that the HH06 shared a very high similarity with many Chinese IBV isolates, however, it had a very low sequence identity with the IBV vaccine strain H120, although IBV HH06 was isolated from the suspected young chicken flock that had been vaccinated with IBV H120. The emerging of IBV HH06 supports the notion that variant IBV isolates can be obtained from vaccinated birds because cross neutralization does not occur between different serotypes. Because both sequence comparison and phylogenetic tree analysis showed that HH06 had a distant relation to H120, we believe that the local prevalence of HH06 may be attributed to the failure of traditional viral vaccine. In addition, our animal experiments show that kidney injury is one of the serious signs of IBV-infected chicken.
The M protein of coronaviruses is a structural membrane protein and plays an important role in the viral assembly process. Here, we have cloned the full-length M gene of the Chinese IBV isolate HH06 and determined its nucleotide sequence. Sequence comparison with other reference strains, which was revealed that the M gene was conserved among various IBV strains inferred from their high sequence similarity, and can be used as a target to develop a nucleic acid-based test for the diagnosis of IBV infection. Many aspects of IBV have been extensively studied [27–32]. Particularly, it is documented that IBV M protein is indispensable for many biological functions including viral core stability [10, 27, 33–36]. In the future, we will express the M gene to further analyze its biological function of the M protein and develop optimal reagents for serological diagnosis.
There is evidence that IBV recombination occurs in the envelope and membrane genes [37]. In this study, many Chinese isolates, such as SAIBK, partridge/GD/S14/2003, CK/CH/LGD/04II, CK/CH/LSC/99I, LDT3, and our newly isolated strain, HH06, were isolated from rather different places in China had very high sequence identity. Our data indicate that the spread of IBV to different regions of China may occur, and whether there is a recombination needs to be investigated in the future.
To further elucidate the molecular evolution of IBV, a phylogenetic tree was constructed according to the M gene sequences. In agreement with the sequence comparison, the tree topology showed the IBV isolates have been divided into several branches, and most Chinese isolates including HH06 isolate were closely clustered in the phylogenetic tree. Most American IBV isolates were also located in neighbored branches. Interestingly, many Chinese IBV isolates that were from different places shared the same branch. For example, SAIBK isolated from Sichuan Province, partridge/GD/S14/2003 and LDT3 isolated from Guangdong Province, and HH06 isolated in Northern China shared the same branch and displayed closer relationships. This result, together with the sequence comparison, allows us to hypothesize that some Chinese IBV isolates may have a common ancestor, although they were isolated from the different regions in mainland China. Particularly, it should be noted that the IBV LDT3 is a peafowl isolate [19, 36]. However, the distinctive dissimilarity between many Chinese isolates and the widely used IBV vaccine strain such as H120 and H52 also indicates that the antigenic drift is likely to occur among some Chinese IBV strains under the long-term immune pressure.
Our results also highlight the importance of developing in time new IBV vaccines against the IBV variants. However, more data and necessary experiments are required to verify our hypothesis and to delineate why the Chinese isolates (HK and Jilin) locate in other different branches in the phylogenetic tree, respectively. In addition, to really understand the relationships among the IBV isolates, more extensive sequencing of the whole genome, the regional vaccination background, and the epidemiology should be investigated in the future.
References
C.C. Tseng, N.Z. Li, C.H. Yao, C.H. Wang, J. Chin. Soc. Vet. Sci. 22, 113–120 (1996)
M. Mase, K. Tsukamoto, K. Imai, S. Yamaguchi, Arch. Virol. 149, 2069–2078 (2004)
P. Britton, S. Evans, B. Dove, M. Davies, R. Casais, D. Cavanagh, J. Virol. Methods 123, 203–211 (2005). doi:https://doi.org/10.1016/j.jviromet.2004.09.017
R. Casais, M. Davies, D. Cavanagh, P. Britton, J. Virol. 79, 8065–8078 (2005). doi:https://doi.org/10.1128/JVI.79.13.8065-8078.2005
R. Casais, B. Dove, D. Cavanagh, P. Britton, J. Virol. 77, 9084–9089 (2003). doi:https://doi.org/10.1128/JVI.77.16.9084-9089.2003
A.A.F. de Vries, M.C. Horzinek, P.J.M. Rottier, R.J. de Groot, Semin. Virol. 8, 33–48 (1997). doi:https://doi.org/10.1006/smvy.1997.0104
M.M. Lai, D. Cavanagh, Adv. Virus Res. 48, 1–100 (1997). doi:https://doi.org/10.1016/S0065-3527(08)60286-9
S.G. Siddell, The Coronaviridae (Plenum Press, New York, NY, 1995), pp. 1–10
K. Mardani, A.H. Noormohammadi, P. Hooper, J. Ignjatovic, G.F. Browning, J. Virol. 82, 2013–2024 (2008). doi:https://doi.org/10.1128/JVI.01694-07
D. Cavanagh, J. Gen. Virol. 64, 1187–1191 (1983). doi:https://doi.org/10.1099/0022-1317-64-5-1187
A.M. Gibertoni, Mde.F. Montassier, J.A. Sena, P.E. Givisiez, C.R. Furuyama, H.J. Montassier, J. Clin. Microbiol. 43, 1982–1984 (2005). doi:https://doi.org/10.1128/JCM.43.4.1982-1984.2005
P.J.M. Rottier, The Coronavirus Membrane Protein (Plenum Press, New York, 1995), pp. 115–139
J. Krijnse Locker, G. Griffith, M.C. Horzinek, P.J.M. Rottier, J. Biol. Chem. 267, 14094–14101 (1992)
J. Krijnse Locker, D.J.E. Opestelten, M. Ericsson, M.C. Horzinek, P.J.M. Rottier, J. Biol. Chem. 270, 8815–8821 (1995). doi:https://doi.org/10.1074/jbc.270.15.8815
B. Charley, H. Laude, J. Virol. 62, 8–11 (1988)
H. Laude, J. Gelfi, L. Lavenant, B. Charley, J. Virol. 66, 743–749 (1992)
P. Baudoux, C. Carrat, L. Besnardeau, B. Charley, H. Laude, J. Virol. 72, 8636–8643 (1998)
S.W. Liu, X.G. Kong, Avian Pathol. 33, 321–327 (2004). doi:https://doi.org/10.1080/0307945042000220697
S.W. Liu, J.F. Chen, Z.X. Han, Q.X. Zhang, Y.H. Shao, X.G. Kong et al., Avian Pathol. 35, 394–399 (2006). doi:https://doi.org/10.1080/03079450600920984
S.W. Liu, Q.X. Zhang, J.D. Chen, Z.X. Han, X. Liu, L. Feng et al., Arch. Virol. 151, 1133–1148 (2006). doi:https://doi.org/10.1007/s00705-005-0695-6
L. Yu, Y. Jiang, S. Low, Z. Wang, S.J. Nam, W. Liu et al., Avian Dis. 45, 416–424 (2001). doi:https://doi.org/10.2307/1592981
S.P. Mondal, C.J. Cardona, Virus Genes 34, 327–341 (2007). doi:https://doi.org/10.1007/s11262-006-0014-7
D.G. Higgins, P.M. Sharp, Gene 73, 237–244 (1988). doi:https://doi.org/10.1016/0378-1119(88)90330-7
B.W. Calnek, Diseases of Poultry, 10th edn. (Iowa State University Press, Ames, IO, 1997)
J.Y. Zhou, D.Y. Zhang, J.X. Ye, L.Q. Cheng, J. Vet. B Med. Infect. Dis. Vet. Public Health 51, 147–152 (2004). doi:https://doi.org/10.1111/j.1439-0450.2004.00744.x
D.X. Ma, L. Pan, X.F. Ren, J.X. Wang, G.X. Li, Vet. Sci. China 37, 926–931 (2007) (in Chinese)
D. Cavanagh, P. Davis, J. Cook, D. Li, Adv. Exp. Med. Biol. 276, 369–372 (1990)
B. Schultze, D. Cavanagh, G. Herrler, Virology 189, 792–794 (1992). doi:https://doi.org/10.1016/0042-6822(92)90608-R
B. Schultze, L. Enjuanes, D. Cavanagh, G. Herrler, Adv. Exp. Med. Biol. 342, 305–310 (1993)
D. Cavanagh, Avian Pathol. 34, 439–448 (2005). doi:https://doi.org/10.1080/03079450500367682
M.W. Jackwood, D.A. Hilt, S.M. Williams, P. Woolcock, C. Cardona, R. O’Connor, Avian Dis. 51, 527–533 (2007). doi:https://doi.org/10.1637/0005-2086(2007)51[527:MASCPA]2.0.CO;2
D. Cavanagh, R. Casais, M. Armesto, T. Hodgson, S. Izadkhasti, M. Davies et al., Vaccine 25, 5558–5562 (2007). doi:https://doi.org/10.1016/j.vaccine.2007.02.046
D. Cavanagh, P.J. Davis, J. Gen. Virol. 69, 621–629 (1988). doi:https://doi.org/10.1099/0022-1317-69-3-621
L.J. Saif, Vet. Microbiol. 37, 285–297 (1993). doi:https://doi.org/10.1016/0378-1135(93)90030-B
E. Corse, C.E. Machamer, J. Virol. 74, 4319–4326 (2000). doi:https://doi.org/10.1128/JVI.74.9.4319-4326.2000
S. Liu, J. Chen, J. Chen, X. Kong, Y. Shao, Z. Han et al., J. Gen. Virol. 86, 719–725 (2005). doi:https://doi.org/10.1099/vir.0.80546-0
J.E. Brooks, A.C. Rainer, R.L. Parr, P. Woolcock, F. Hoeff, E.W. Collisson, Virus Res. 100, 191–198 (2004). doi:https://doi.org/10.1016/j.virusres.2003.11.016
Acknowledgments
We thank the anonymous reviewers and the editors of this journal for their critical review on the paper. The authors’ research works were supported by funds from Heilongjiang Provincial Science and Technology Department (LC06C01) and Harbin Science and Technology Bureau (2006RFLXN004), National Natural Science Foundation of China (30700590; 30700591), Heilongjiang Provincial Education Department SARS Project (2003fz012), Ministry of Education of China (NO706019) and Program for Innovative Research Team of Northeast Agricultural University (CXZ008-1).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ren, X., Yin, J., Ma, D. et al. Characterization and membrane gene-based phylogenetic analysis of avian infectious bronchitis virus Chinese strain HH06. Virus Genes 38, 39–45 (2009). https://doi.org/10.1007/s11262-008-0280-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0280-7