
Abstract
Lumpy skin disease (LSD) is a devastating viral disease of cattle which has recently spread from Africa into the countries of the Middle East. The aim of the present study was to investigate the relationships among lumpy skin disease viruses (LSDV) isolated from different regions of Iran and the origin and spread of these viruses. In this study, a total of 234 blood samples from clinically affected animals from four provinces in the northwest of Iran were screened for LSDV using polymerase chain reaction (PCR). From 80 positive samples for LSDV detected by PCR, the partial P32 gene (759 bp) of 12 isolates were sequenced and phylogenetically analyzed. LSD viruses were grouped in three subclusters with an overall 97.1–100% nucleotide identity. LSDVs isolated from Gilan showed lowest nucleotide identity with the other LSDVs. Four isolates of LSDV including KO-1, EA-1, EA-3, and WA-3 showed 100% similarity with each other and also with the Neethling strain. Phylogenetic analysis indicated that the identified LSDVs were closely related to each other and had high-sequence homology with other LSDV isolates from Africa. It was concluded that LSD outbreak probably occurred in the northwest of Iran by LSDVs entering the country from Iraq and P32 nucleotide sequence information obtained in the present study is a valuable resource in understanding the genetic nature and molecular epidemiology of local LSDV isolates which can be used for future vaccine development based on the circulating strains in the region.
Similar content being viewed by others


Introduction
Lumpy skin disease (LSD) is a major infectious disease of cattle, causing mild to severe symptoms in infected animals (Abera et al. 2015; Tuppurainen and Oura 2012; Tuppurainen et al. 2017). The disease is of great economic importance in both beef and dairy industries (Kumar 2011) due to its impact on the reduction in milk production, abortion, infertility, loss of condition, and damage to the hide (Tuppurainen et al. 2017). The causative agent of LSD is lumpy skin disease virus (LSDV), a DNA virus of the Poxviridae family and of the genus Capripoxvirus (CaPV). LSDV is closely related to the other two viruses of the genus Capripoxvirus including sheep pox virus (SPPV) and goat pox virus (GTPV) with high antigenic resemblance and genome identities of at least 96% (Babiuk et al. 2008).
LSD is widespread across Africa and occurs at regular intervals in endemic areas causing particularly severe outbreaks in the horn of Africa (Tuppurainen et al. 2017). Until 2012, LSD occurred sporadically in the Middle East region. However, since 2012, outbreaks of LSD have been reported in Israel, Syria, Lebanon, Jordan, Turkey, Iraq, Iran, Azerbaijan, Kuwait, and Saudi Arabia (Al-Salihi and Hassan 2015; Ben-Gera et al. 2015; Kasem et al. 2018; Mercier et al. 2018; Sameea Yousefi et al. 2016; Sevik and Dogan 2017). Recently, LSD has been aggressively spreading in the European Union, with its first incursions reported in Greece, Russia, Dagestan, and Chechnya despite excessive vaccination campaigns carried out in these regions, raising concerns that the disease will continue to spread to Europe and Asia (Agianniotaki et al. 2017b; Salnikov et al. 2018; Tuppurainen et al. 2014; Tuppurainen et al. 2017).
Conventional serological methods cannot distinguish SPPV, GTPV, and LSDV. Thus, the most commonly used technique for differentiating these viruses is PCR (Zhou et al. 2012). Restriction endonuclease analysis (REA) of the genomic DNA of SPPV, GTPV, and LSDV viruses has earlier shown that these viruses are genetically closely related but are barely distinguishable. However, according to the OIE (2016), REA is not always a reliable technique due to recombination events in the viral genome. Recently, a duplex real-time PCR method has been used for the specific detection and differentiation of both wild-type LSDV and Neethling/SIS vaccine viruses (Agianniotaki et al. 2017a). Furthermore, genomic analyses of SPPV, GTPV, and LSDV viruses have revealed that they share considerable homology with each other (Tulman et al. 2002); however, genome sequencing has shown that CaPVs are phylogenetically distinct (Lamien et al. 2011; Le Goff et al. 2009). Therefore, sequence information and phylogenetic analysis can be used in differentiating and elucidating the genetic relatedness of these viruses (Zhou et al. 2012).
The P32 protein is a highly antigenic structural protein of all strains of CaPVs which has been used in serological detection of these viruses (Carn et al. 1994; Heine et al. 1999; OIE 2016; Tian et al. 2010). Therefore, P32 has been reported as an important antigen in the pathogenicity and diagnosis of CaPVs (Tian et al. 2010). In a number of studies, P32 gene has been used as a candidate gene for detection and molecular characterization of capripoxviruses (El-Kholy et al. 2008; Hosamani et al. 2004; Mafirakureva et al. 2017; Zhao et al. 2017). In a recent study by Mafirakureva et al. (2017), it was reported that phylogenetic analysis of LSDV isolates based on P32 gene could be a reliable tool in determining the origin of the LSDV isolates.
In 2014, the outbreaks of LSD have been reported in northwestern provinces of Iran, causing major economic losses due its mortality and reduction in milk production (Sameea Yousefi et al. 2016). Because of high antigenic homology and cross-protection between sheep pox, goat pox, and LSD viruses (Kitching 2003), attenuated SPPV and GTPV vaccines, such as KSGP O-240, Yugoslavian RM65, and Romanian SPPV strains, have been used against LSDV in Iran. Since there was no information about the genotypic nature of the LSDVs circulating in the region and the neighboring countries, it was of great importance to reveal the origin and relatedness of LSDV isolates with those in the other countries. The present study was designed to investigate the genetic nature of circulating LSDVs in the regions where outbreaks of LSD occurred and to determine the phylogenetic relatedness of the isolated LSDVs. The P32 gene of a number of 12 detected LSDVs were amplified and sequenced. The results revealed the circulation of LSDVs in the northwest of Iran, which were genetically related to each other by more than 99% identity.
Materials and methods
Blood sample collection
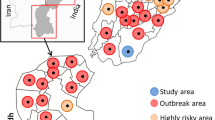
During the outbreak of LSD between 2014 and 2015 in different regions of Iran, blood samples were collected from 234 cattle (122 clinically diseased and 112 nonclinically diseased) from four provinces including West Azerbaijan, East Azerbaijan, Kurdistan, and Gilan as it was reported in our previous study (Sameea Yousefi et al. 2016). An amount of whole blood (3 ml) was collected in EDTA-coated tube from each animal. Blood samples were immediately transferred to the central laboratory at the Faculty of Veterinary Medicine, Urmia University. Blood samples were kept at − 20 °C until they were used for viral DNA extraction.
DNA extraction
Total DNA was extracted from 200 μl of blood samples using a NucleoSpin® Blood Kit (Macherey-Nagel, Germany) according to the instructions specified by the kit manufacturer. Extracted DNA was quantified using NanoDrop 2000c (Thermo Scientific, USA).
PCR
PCR-based detection of LSDVs was performed using two primer pairs described by Tageldin et al. (2014). The first pair of primers, including DW-TK (5′-GCC GAT AAC ATA TATAGA CCC-3′) and OP49 (5′-GTG CTA TCT AGTGCA GCTAT-3′), was used to amplify a 434-bp fragment from all CaPVs, and the second pair of primers, consisting of L132F (5′-CAC TTC CCT TTTAAGC-3′) and L132R (5′-CAT TCTACA ATC TCC ATGCG-3′), was used to amplify a 492-bp fragment which was specific for LSDV. The initial primer pair used for virus identification is homologous to the regions of LSDV thymidine kinase (TK) gene, which amplifies genomic fragment between positions 56698–57132. The second primer pair binds to the ORF132 gene which is unique to LSDV. The PCR reactions were prepared in a total volume of 25 μl mixtures containing 50–100 ng genomic DNA; 0.5 μM of each primer; 50 μM of each ATP, CTP, GTP, and TTP, 2 mM MgCl2; 2.5 units of SmarTaq DNA polymerase (SinaClon, Iran); and 2.5 μl 10× PCR buffer. The PCR reactions were performed using a QB Cycler® gradient thermal cycler (Quanta Biotech, England) under the following thermal condition: initial denaturation for 90 s at 95 °C, followed by 35 cycles of denaturation (45 s at 95 °C), primer annealing (45 s at 56 °C), and strand extension (60 s at 72 °C), ending with a final strand extension step for 7 min at 72 °C. These conditions were used for both primer pairs (Tageldin et al. 2014). The resultant PCR products were separated in 1.5% (w/v) agarose gel containing SimplySafe (EURx, Poland) 2.5 μl/50 ml gel for 1 h at 75 V and visualized under UV transilluminator.
For phylogenetic analysis, the third set of primers was designed based on the P32 gene of the CaPVs sequences retrieved from GenBank. The third set of designed primers, including P32-F (5′-TCGTTGGTCGCGAAATTTCAG-3′) and P32-R (5′-GAGCCATCCATTTTCCAACTCT-3′), targeted a fragment of 759 bp in size between positions 65163 and 65921 based on LSDV strain NI-2490 genome (accession no. AF325528.1). The PCR reaction was prepared as previously described. Amplification was performed using 35 cycles of incubation at 95 °C for 45 s, 56 °C for 45 s, and 72 °C for 65 s, with a final extension at 72 °C for 5 min.
Purification of PCR products
PCR products were purified using a gel purification kit (Gent Bio, Taiwan) in line with the manufacturer’s instructions.
Nucleotide sequencing and phylogenetic analysis
Purified P32 gene PCR products from each LSDV and two vaccines which are used against LSD in Iran (SPPV and GPV vaccines) along with P32-F primer were sent to Macrogen Company (South Korea) for nucleotide sequencing using BigDye v.3.1 chemistry in 23 ABI 3730XLs genetic analyzer. Nucleotide sequences of P32 genes from LSDVs were searched in the GenBank database (National Centre for Biotechnology Information, Rockville Pike, Bethesda, MD) using the advanced BLAST similarity search option (available at http://www.ncbi.nlm.nih.gov) discovering whether the obtained sequences belonged to CaPV genus. Nucleotide sequences from the present study were aligned and compared with the P32 gene sequences of other LSDV, SPPV, and GPV viruses retrieved from GenBank using Clustal W. A phylogenetic tree was generated using the maximum-likelihood method based on the Tamura-Nei model (Tamura and Nei 1993) in MEGA v.6.0 software (Tamura et al. 2013). Estimates of similarities between sequences were also calculated using the maximum composite likelihood model (Tamura et al. 2004). Nucleotide sequences of P32 gene for all 12 LSDV isolates were submitted to the GenBank database under accession numbers KX960769–KX960782.
Results
Field LSDVs
From a total of 234 screened cattle for LSDV, 80 (34.1%) cattle were positive using PCR test. A number of 12 detected LSDVs from examined cattle, belonging to four northwestern provinces including Gilan (four isolates SH-1, SH-2, SH-3, and SH-4), West Azerbaijan (three isolates WA-1, WA-2, and WA-3), East Azerbaijan (four isolates EA-1, EA-2, EA-3, and EA-4), and Kurdistan (one isolate KO-1), were chosen for phylogenetic analysis.
PCR
A fragment of 434 bp in size was amplified from all LSDVs using the first pair of primers confirming the genus Capripoxvirus (Fig. 1). Amplification of the 494-bp PCR products using the second pair of primers was unsuccessful for a number of isolates (data not shown). Therefore, the third pair of primers targeting P32 gene was designed and a fragment of 759 bp in size of P32 gene was amplified from all LSDVs and two SPPV and GTPV vaccines (Fig. 2).

Amplification of a fragment of 434 bp in size from a number of examined cattle. PCR products were run in 1.5% agarose gel. Lane 1, molecular ladder 100 bp (SinaClon, Iran); lanes 2–4, positive samples (SH-1, WA-1, and EA-1) from examined cattle; lane 5, positive control (SPPV vaccine); lane 6, negative control

Amplification of a fragment of 759 bp in size from three examined cattle. PCR products were run in 1.5% agarose gel. Lane 1, molecular ladder 100 bp (SinaClon, Iran); lanes 2–4, positive samples (SH-1, WA-1, and EA-1) from examined cattle
Phylogenetic analysis and estimated identity between P32 gene sequences of CaPVs
A phylogenetic tree was constructed based on the nucleotide sequences of P32 gene (717 bp) of CaPVs. Since there was a limited number of P32 gene nucleotide sequences corresponding to the region we sequenced in the GenBank, we used P32 gene nucleotide sequences from SPPV and GTPV viruses in phylogenetic analysis. LSDV, SPPV, and GTPV viruses were clustered in distinct and separate clusters, confirming the genotypic differences among these three members of CaPVs genus in spite of high conserve nature of their genome. All 12 viruses detected from examined cattle in the present study were closely clustered with LSDV reported from other parts of the world. LSDVs in the generated phylogenetic tree were grouped into three subclusters. Subcluster I-a had four isolates from three neighboring provinces (West and East Azerbaijan and Kurdistan) and two isolates from Kenya and South Africa, subcluster I-b had all isolates from Gilan Province and one isolate from West Azerbaijan, and subcluster I-c included two isolates from East and West Azerbaijan and the Neethling strain from South Africa (Fig. 3). LSDVs from Gilan were closely clustered with each other, almost far from other LSDVs that belonged to other provinces.

Phylogenetic analysis of LSDVs detected in the present study and CaPVs (LSDV, SPPV, and GTPV) from GenBank by maximum likelihood method in MEGA6. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 29 nucleotide sequences. Codon positions were first + second + third + noncoding. All positions containing gaps and missing data were eliminated
The estimated sequence identity between P32 sequences of CaPVs revealed an overall 92.6–100% nucleotide resemblance among these viruses. LSDV isolates and two SPPV and GTPV vaccines in the present study showed 96.7–100% similarity in their P32 gene nucleotide sequences. The P32 nucleotide similarity percentage between LSDVs from Iran and other countries varied from 97.1 to 100%. LSDVs from Gilan (SH-1, SH-2, SH-3, and SH-4) showed the lowest identity with other LSDVs detected in this study. Interestingly, P32 gene of four LSDVs including KO-1, EA-1, EA-3, and WA-3 showed 100% similarity with each other and also with the Neethling strain of LSDV from Kenya and South Africa (Table 1).
Discussion
Detailed knowledge of viral genomes is recently used for vaccine design strategies since such information allows researchers to analyze the origin and conservation of genes among the circulating LSDV at a certain region (Tuppurainen et al. 2017). Since the northwest of Iran has common borders with four countries including Iraq, Turkey, Armenia, and the Republic of Azerbaijan, investigating the origins of LSDV is of great epidemiological importance. In the present study, cattle with and without LSD symptoms were investigated for the presence of LSDV using PCR. While a number of 80 cattle were positive for LSDVs in the initial PCR, the second PCR, which was employed to detect specifically LSDVs, failed to detect LSDV in a number of positive samples. Low specificity of employed primers for Iranian LSDV could be the reason for this PCR failure. A number of 12 detected LSDVs from the examined animals were further characterized based on their P32 gene nucleotide sequences. Phylogenetic tree and sequence identity analyses were used to evaluate the genetic nature of detected LSDVs in cattle from the northwest of Iran, where disease outbreak was reported for the first time. Before the outbreak of LSD in Iran, the disease was reported by OIE in Iraq. Therefore, as it was speculated in our previous work (Sameea Yousefi et al. 2016), LSD could have entered Iran by the uncontrolled movements of infected animals between the two countries.
Despite high-sequence homology between CaPVs (Tulman et al. 2002) alignment and phylogenetic analysis of P32 gene sequences, grouped LSDVs, SPPVs, and GTPVs in three distinct clusters confirm the findings of previous studies (Hosamani et al. 2004; Mafirakureva et al. 2017; Zhou et al. 2012). In LSDV cluster, all isolates belonging to Gilan Province were closely subclustered with each other far from other LSDVs (except for one isolate WA-1) from other provinces, revealing the possibility of different genetic nature of the virus even in the same country. Based on the phylogenetic analysis of P32 gene, most of the field strains of LSDVs clustered far from LSDV-Neethling vaccine, indicating the possibility of using this gene as a candidate for differentiating vaccine strains of LSDV from field isolates. Furthermore, comparative analyses of the phylogenetic trees constructed based on GPCR (Gelaye et al. 2015; Le Goff et al. 2009; Salnikov et al. 2018; Su et al. 2015; Zhou et al. 2012), RPO30 (Lamien et al. 2011; Su et al. 2015), and P32 genes (Mafirakureva et al. 2017) of LSDV demonstrate that P32 gene was a better candidate gene for illustrating genetic variation between LSDVs.
Recently, the sequencing of GPCR and P32 genes has been used to identify and characterize LSDVs from SPPV and GTPV (Mafirakureva et al. 2017; Salnikov et al. 2018). Phylogenetic analysis of different LSDV isolates using P32 gene revealed two subclusters in the examined LSDVs (Mafirakureva et al. 2017), generating a better picture of differences between LSDVs than the phylogenetic analysis based on GPCR gene in which all LSDVs clustered together (Salnikov et al. 2018). In the present study, examined LSDVs were divided into three subclusters, which it was due to the sequencing of a larger part of P32 gene (717 bp) in comparison with Mafirakureva et al.’s work (2017), in which they sequenced a smaller part of the P32 gene (192 bp).
Nucleotide sequence similarity data showed that the identified LSDVs from three different provinces were identical. Moreover, these isolates enjoyed 100% similarity with two other LSDV strains from Kenya and South Africa, revealing a high conserved nature of LSDV genome despite virus circulation in different geographical locations. However, phylogenetic tree constructed based on P32 gene of LSDVs from the northwestern region of Iran showed that LSDVs from Gilan Province (SH-1, SH-2, SH-3, and SH-4) were clustered separately from the other LSDVs isolated from neighboring provinces in Iran. Phylogenetic analysis of the 12 LSDV isolates based on the nucleotide sequence of the P32 gene showed that these 12 isolates could be grouped into three distinct subclusters. Subcluster I-a had the Iranian isolates from three different provinces, while all LSDV isolates from Gilan were placed in the subcluster I-b. This reveals that different strains of LSDV could be in circulation in Iran. This finding is congruent with those reported by Mafirakureva et al. (2017).
In a previous study by Stram et al. (2008), a fragment of 466 bp in size of LSDV genome termini was used for the detection and phylogenetic analysis of CaPVs. They showed that the constructed phylogenetic tree was able to distinguish among various CaPVs but failed to differentiate different isolates even though the surveyed region is one of the most divergent regions of the viral genome. However, in our study, the generated phylogenetic tree based on P32 gene was able to not only differentiate different CaPVs but also roughly distinguish LSDVs originated from one of the examined regions. This finding reveals that P32 gene is a better marker than LSDV genome termini for distinguishing between different CaPVs and isolates.
Conclusion
In conclusion, this study found that LSDV keeps circulating in different provinces in Iran. Moreover, as described in this study, P32 nucleotide sequences of LSDV provide insights regarding the genotypic nature of circulating LSDV strains in the northwest of Iran for the first time. This information could be of paramount importance for tracking the origins of the viruses and future studies and efforts on vaccine development against LSDV.
References
Abera, Z., Degefu, H., Gari, G., and Ayana, Z., 2015. Review on Epidemiology and Economic Importance of Lumpy Skin Disease. International Journal of Basic and Applied Virology, 4, 8–21
Agianniotaki, E.I., Chaintoutis, S.C., Haegeman, A., Tasioudi, K.E., De Leeuw, I., Katsoulos, P.D., Sachpatzidis, A., De Clercq, K., Alexandropoulos, T., Polizopoulou, Z.S., Chondrokouki, E.D., and Dovas, C.I., 2017a. Development and validation of a TaqMan probe-based real-time PCR method for the differentiation of wild type lumpy skin disease virus from vaccine virus strains. Journal of Virological Methods, 249, 48–57
Agianniotaki, E.I., Tasioudi, K.E., Chaintoutis, S.C., Iliadou, P., Mangana-Vougiouka, O., Kirtzalidou, A., Alexandropoulos, T., Sachpatzidis, A., Plevraki, E., Dovas, C.I., and Chondrokouki, E., 2017b. Lumpy skin disease outbreaks in Greece during 2015-16, implementation of emergency immunization and genetic differentiation between field isolates and vaccine virus strains. Veterinary Microbiology, 201, 78–84
Al-Salihi, K.A., and Hassan, I.Q., 2015. Lumpy Skin Disease in Iraq: Study of the Disease Emergence. Transboundary and Emerging Diseases, 62, 457–462
Babiuk, S., Bowden, T.R., Boyle, D.B., Wallace, D.B., and Kitching, R.P., 2008. Capripoxviruses: an emerging worldwide threat to sheep, goats and cattle. Transboundary and Emerging Diseases, 55, 263–272
Ben-Gera, J., Klement, E., Khinich, E., Stram, Y., and Shpigel, N.Y., 2015. Comparison of the efficacy of Neethling lumpy skin disease virus and x10RM65 sheep-pox live attenuated vaccines for the prevention of lumpy skin disease - The results of a randomized controlled field study. Vaccine, 33, 4837–4842
Carn, V.M., Kitching, R.P., Hammond, J.M., and Chand, P., 1994. Use of a recombinant antigen in an indirect ELISA for detecting bovine antibody to capripoxvirus. Journal of Virological Methods, 49, 285–294
El-Kholy, A.A., Soliman, H.M.T., and Abdelrahman, K.A., 2008. Polymerase chain reaction for rapid diagnosis of a recent lumpy skin disease virus incursion to Egypt. Arab Journal of Biotechnology, 11, 293–302
Gelaye, E., Belay, A., Ayelet, G., Jenberie, S., Yami, M., Loitsch, A., Tuppurainen, E., Grabherr, R., Diallo, A., and Lamien, C.E., 2015. Capripox disease in Ethiopia: Genetic differences between field isolates and vaccine strain, and implications for vaccination failure. Antiviral Research, 119, 28–35
Le Goff, C., Lamien, C.E., Fakhfakh, E., Chadeyras, A., Aba-Adulugba, E., Libeau, G., Tuppurainen, E., Wallace, D.B., Adam, T., Silber, R., Gulyaz, V., Madani, H., Caufour, P., Hammami, S., Diallo, A., and Albina, E., 2009. Capripoxvirus G-protein-coupled chemokine receptor: a host-range gene suitable for virus animal origin discrimination. The Journal of General Virology, 90, 1967–1977
Heine, H.G., Stevens, M.P., Foord, A.J., and Boyle, D.B., 1999. A capripoxvirus detection PCR and antibody ELISA based on the major antigen P32, the homolog of the vaccinia virus H3L gene. Journal of Immunological Methods, 227, 187–196
Hosamani, M., Mondal, B., Tembhurne, P.A., Bandyopadhyay, S.K., Singh, R.K., and Rasool, T.J., 2004. Differentiation of sheep pox and goat poxviruses by sequence analysis and PCR-RFLP of P32 gene. Virus Genes, 29, 73–80
Kasem, S., Saleh, M., Qasim, I., Hashim, O., Alkarar, A., Abu-Obeida, A., Gaafer, A., Hussien, R., Al-Sahaf, A., Al-Doweriej, A., Bayoumi, F., Hodhood, A., and Abdelatif, M., 2018. Outbreak investigation and molecular diagnosis of Lumpy skin disease among livestock in Saudi Arabia 2016. Transboundary and Emerging Diseases, 65, 494–500
Kitching, R.P., 2003. Vaccines for lumpy skin disease, sheep pox and goat pox. Developments in Biologicals, 114, 161–167
Kumar, S.M., 2011. An Outbreak of Lumpy Skin Disease in a Holestein Dairy Herd in Oman: A Clinical Report. Asian Journal of Animal and Veterinary Advances, 6, 851–859
Lamien, C.E., Le Goff, C., Silber, R., Wallace, D.B., Gulyaz, V., Tuppurainen, E., Madani, H., Caufour, P., Adam, T., El Harrak, M., Luckins, A.G., Albina, E., and Diallo, A., 2011. Use of the Capripoxvirus homologue of Vaccinia virus 30 kDa RNA polymerase subunit (RPO30) gene as a novel diagnostic and genotyping target: development of a classical PCR method to differentiate Goat poxvirus from Sheep poxvirus. Veterinary Microbiology, 149, 30–39
Mafirakureva, P., Saidi, B., and Mbanga, J., 2017. Incidence and molecular characterisation of lumpy skin disease virus in Zimbabwe using the P32 gene. Tropical Animal Health and Production, 49, 47–54
Mercier, A., Arsevska, E., Bournez, L., Bronner, A., Calavas, D., Cauchard, J., Falala, S., Caufour, P., Tisseuil, C., Lefrancois, T., and Lancelot, R., 2018. Spread rate of lumpy skin disease in the Balkans, 2015-2016. Transboundary and Emerging Diseases, 65, 240–243
OIE, 2016. Lumpy Skin Disease. In: OIE (ed), Biomed Res Int, 2016, OIE Terrestrial Manual.
Salnikov, N., Usadov, T., Kolcov, A., Zhivoderov, S., Morgunov, Y., Gerasimov, V., Gogin, A., Titov, I., Yurkov, S., Malogolovkin, A., Kolbasov, D., and Lunitsyn, A., 2018. Identification and characterization of lumpy skin disease virus isolated from cattle in the Republic of North Ossetia-Alania in 2015. Transboundary and Emerging Diseases,
Sameea Yousefi, P., Mardani, K., Dalir-Naghadeh, B., and Jalilzadeh-Amin, G., 2016. Epidemiological study of lumpy skin disease outbreaks in north-western Iran. Transboundary and Emerging Diseases, 64, 1782–1789
Sevik, M., and Dogan, M., 2017. Epidemiological and molecular studies on lumpy skin disease outbreaks in Turkey during 2014-2015. Transboundary and Emerging Diseases, 64, 1268–1279
Stram, Y., Kuznetzova, L., Friedgut, O., Gelman, B., Yadin, H., and Rubinstein-Guini, M., 2008. The use of lumpy skin disease virus genome termini for detection and phylogenetic analysis. Journal of Virological Methods, 151, 225–229
Su, H.L., Jia, H.J., Yin, C., Jing, Z.Z., Luo, X.N., and Chen, Y.X., 2015. Phylogenetic analysis of Gansu sheeppox virus isolates based on P32, GPCR, and RPO30 genes. Genetics and Molecular Research, 14, 1887–1898
Tageldin, M.H., Wallace, D.B., Gerdes, G.H., Putterill, J.F., Greyling, R.R., Phosiwa, M.N., Al Busaidy, R.M., and Al Ismaaily, S.I., 2014. Lumpy skin disease of cattle: an emerging problem in the Sultanate of Oman. Tropical Animal Health and Production, 46, 241–246
Tamura, K., and Nei, M., 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution, 10, 512–526
Tamura, K., Nei, M., and Kumar, S., 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S., 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729
Tian, H., Chen, Y., Wu, J., Shang, Y., and Liu, X., 2010. Serodiagnosis of sheeppox and goatpox using an indirect ELISA based on synthetic peptide targeting for the major antigen P32. Virology Journal, 7, 245
Tulman, E.R., Afonso, C.L., Lu, Z., Zsak, L., Sur, J.H., Sandybaev, N.T., Kerembekova, U.Z., Zaitsev, V.L., Kutish, G.F., and Rock, D.L., 2002. The genomes of sheeppox and goatpox viruses. Journal of Virology, 76, 6054–6061
Tuppurainen, E.S., and Oura, C.A., 2012. Review: lumpy skin disease: an emerging threat to Europe, the Middle East and Asia. Transboundary and Emerging Diseases, 59, 40–48
Tuppurainen, E.S., Pearson, C.R., Bachanek-Bankowska, K., Knowles, N.J., Amareen, S., Frost, L., Henstock, M.R., Lamien, C.E., Diallo, A., and Mertens, P.P., 2014. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antiviral Research, 109, 1–6
Tuppurainen, E.S., Venter, E.H., Shisler, J.L., Gari, G., Mekonnen, G.A., Juleff, N., Lyons, N.A., De Clercq, K., Upton, C., Bowden, T.R., Babiuk, S., and Babiuk, L.A., 2017. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transboundary and Emerging Diseases, 64, 729–745
Zhao, Z., Wu, G., Yan, X., Zhu, X., Li, J., Zhu, H., Zhang, Z., and Zhang, Q., 2017. Development of duplex PCR for differential detection of goatpox and sheeppox viruses. BMC Veterinary Research, 13, 278
Zhou, T., Jia, H., Chen, G., He, X., Fang, Y., Wang, X., Guan, Q., Zeng, S., Cui, Q., and Jing, Z., 2012. Phylogenetic analysis of Chinese sheeppox and goatpox virus isolates. Virology Journal, 9, 25
Acknowledgments
The authors extend their gratitude to Dr. N. Nowruzi, Dr. A. Piryounesi, and M. Alamdari for their assistance in the collection of samples. We are also grateful to Urmia University for funding this project.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sameea Yousefi, P., Dalir-Naghadeh, B., Mardani, K. et al. Phylogenetic analysis of the lumpy skin disease viruses in northwest of Iran. Trop Anim Health Prod 50, 1851–1858 (2018). https://doi.org/10.1007/s11250-018-1634-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-018-1634-3