
Abstract
Avian influenza is a highly contagious respiratory disease of poultry caused by influenza A viruses, family Orthomyxoviridae. H9N2 avian influenza outbreaks are a major problem of the poultry industry in Iran. To determine the genetic differences between field viruses and the vaccine strain, the genomes of four strains isolated in 2011 from vaccinated broiler flocks with a history of respiratory illness were sequenced. Genetic and serological comparisons were made. Sequence analysis of the hemagglutinin (HA) and neuraminidase (NA) genes indicated that the isolated strains shared nucleotide homologies of 91.6–93.9 and 90.2–91.7 % with the vaccine strain, respectively. Phylogenetic analyses of HA and NA genes showed that all strains isolated in this study fell into the same group and belonged to the influenza A virus (A)/quail/Hong Kong/G1/97 H9N2 sublineage. Several amino acids have changed at the antigenic sites in HA in the field viruses. Extra potential glycosylation sites were observed in the HA and NA proteins expressed by the current isolates relative to those in the vaccine strain. The deduced amino acid sequence at the cleavage site of HA in recent isolates is the KSSR/GLF motif, whereas it is RSSR/GLF in the vaccine strain. A serological analysis revealed that the currently circulating strains are antigenically distinct from the vaccine strain. These results suggest that the commercial vaccine is insufficiently genetically and antigenically similar to the viruses currently circulating in the region. These findings confirm that it is important to monitor the genetic and antigenic variations in H9N2 influenza viruses when selecting a vaccine strain.
Similar content being viewed by others

Introduction
Avian influenza (AI) is a highly contagious respiratory disease of poultry caused by influenza A viruses of the family Orthomyxoviridae. A total of 18 hemagglutinin (HA) and 11 neuraminidase (NA) subtypes have so far been identified (Tong et al. 2013). Since 1998, H9N2 avian influenza outbreaks have been one of the major problems of the Iranian poultry industry (Nilli and Assasi 2003; Bashashati et al. 2013). These highly contagious viruses have spread in poultry flocks throughout the different provinces of the country, so a vaccination strategy using an inactivated H9N2 vaccine based on strain A/chicken (Ck)/Iran/1221/1998 was adopted to control AI disease in Iran (Vasfi Marandi and Bozorgmehri Fard 2002). Despite this vaccination program, outbreaks have continued to occur in several broiler flocks, with great economic losses from both mortality and weight loss. Previous studies have reported that many substitutions have occurred in the HA and NA genes of the Iranian H9N2 viruses in recent years (Bashashati et al. 2013; Norouzian et al. 2014), which might affect the pathogenicity and antigenicity of these avian influenza viruses. Therefore, it is important to investigate whether the circulating H9N2 viruses have undergone further significant genetic and antigenic changes with the advent of vaccination. Antigenic and phylogenetic analysis can also be effective approaches to vaccine development (Park et al. 2011; Lee and Song 2013). Therefore, we characterized four H9N2 viruses recently isolated from commercial farms and the vaccine strain both antigenically and phylogenetically.
Material and methods
Three hundred twenty tissue samples were collected from 25 broiler chicken farms in northeast Iran (Ir) between January and December 2011. The tissue samples usually included the trachea and lungs. All the samples were collected from Ross broiler chickens with a history of respiratory symptoms, such as coughing, sneezing, and ocular and nasal discharge. The chickens showed symptoms between 25 and 40 days of age. The lesions typically included congestion and inflammation of the trachea and lungs. These flocks had satisfactory levels of biosecurity, achieved with segregation, cleaning, and disinfection. They were free of other respiratory diseases, including infection bronchitis, Newcastle disease, and mycoplasmosis. Microbiological test results were often negative for bacterial pathogens, but secondary bacterial pathogens were sometimes isolated, such as Escherichia coli. The morbidity was high, and the mortality rate was 10–15 %. The maternal antibodies in the 1-day-old chickens had a hemagglutination inhibition (HI) titer of 24–26, and the vaccination program was performed with an inactivated oil-emulsion vaccine against H9N2 influenza virus administered to 7–10-day-old chickens, when their maternal antibodies are low. Virus was isolated in 10-day-old specific pathogen-free (SPF)-embryonated chicken eggs. The virus subtypes were determined with standard HI and neuraminidase inhibition (NI) tests, as described previously (Alexander and Spackman 1981). The four viruses isolated in this study are shown in Table 1. The A/Ck/Ir/1221/98 H9N2 virus, the strain selected for vaccine production in Iran since 1998, was included in the molecular and phylogenetic analyses. Antigenic characterization was performed with polyclonal antibodies raised in 5-week-old SPF chickens inoculated with A/Ck/Ir/1221/98.
Viral RNA was extracted directly from the allantoic fluid with the High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany), according to the manufacturer’s instructions. Reverse transcription (RT) followed by PCR was performed with specific primers, as described previously (Hoffmann et al. 2001), in a 50-μl reaction mixture containing 10 μl of 5× reaction buffer, 1 μl of mixed dNTPs, 1 μl of AMV reverse transcriptase (Titan One Tube RT–PCR System; Roche Diagnostics), 2 μl of each primer, 4 μl of RNA template, 2.5 μl of dithiothreitol, and 27.5 μl of distilled water. The PCR cycling parameters were 94 °C for 2 min and 40 cycles of 94 °C for 1 min, 51 °C for 1 min, and 68 °C for 1 min, followed by a final extension at 68 °C for 10 min. The PCR products were purified with the High Pure PCR Product Purification Kit (Roche Diagnostics). The PCR products were separated electrophoretically on low-melting point agarose, and the distinct bands were excised from the gel and purified for sequencing (by MWG-Biotech AG, Germany). The nucleotide and deduced amino acid sequences of the HA and NA genes of the newly isolated strains were edited with BioEdit version 7 (Hall 1999) and were aligned with ClustalW. A phylogenetic analysis was performed with the MEGA 5 program (Tamura et al. 2011). The evolutionary relationships based on the HA and NA nucleotide sequences were determined by comparing the Iranian isolates and selected H9N2 viruses isolated in several other countries with established Eurasian H9N2 lineages: the G1-like sublineage, Y280-like sublineage, and Korean-like sublineage, represented by prototype viruses A/quail/Hong Kong/G1/97, A/duck/Hong Kong/Y280/97, and A/chicken/Korea/38349-p96323/96, respectively (Xu et al. 2007). The current vaccine strain was also included in the analysis. Phylogenetic trees were constructed using the neighbor-joining method with maximum composite likelihood. The internal branching probabilities were determined with a bootstrap analysis of 1000 replicates and indicated by percentage values on each branch.
Potential glycosylation sites were predicted as NXS/T motifs, in which X is any amino acid except proline. To investigate the amino acid variations in the vaccine and field viruses, a multisequence alignment of derived HA protein sequences was constructed. The antigenic sites in the H9N2 viruses were identified based on the results of previous studies (Kaverin et al. 2004; Okamatsu et al. 2008; Wan et al. 2014). Identification of the amino acids in the H9 antigenic sites revealed different distributions of antigenic areas among the other subtypes. There are at least two antigenic sites in the H9 HA molecule (Kaverin et al. 2004), and Okamatsu (2008) defined at least five antigenic sites in the HA protein of H9N2. The substitution of some critical amino acids is also related to the antigenicity of the virus (Wan et al. 2014). In total, multiple amino acids at positions 72, 98, 124, 127, 131, 141, 143, 147, 148, 153, 155, 164, 165, 167, 168, 170, 182, 183, 196, 197, 200, 201, 206, 207, 212, and 234 (H9 numbering) are related to the antigenicity of the H9N2 viruses (Kaverin et al. 2004; Okamatsu et al. 2008; Wan et al. 2014).
Accession numbers
The nucleotide sequences determined in this study have been deposited in the GenBank database under accession numbers KF800938–KF800947.
Results
The sequenced parts of the HA genes contained 1720 nucleotides, and those of the NA genes included 1410 nucleotides. The amino acid sequences of the HA proteins of the currently circulating isolates shared 91.6–93.9 % homology with that of the vaccine strain, and the NA proteins shared 90.2–91.7 % homology with the vaccine strain NA protein. The deduced amino acid sequences at the cleavage site of HA contained the KSSR/GLF motif in all the isolates and an RSSR/GLF motif in the vaccine strain. Seven potential glycosylation sites were identified in each HA protein of the currently circulating strains, at positions 29–31 (NST), 105–107 (NGT), 141–143 (NVT), 298–300 (NST), 305–307 (NIS), 492–494 (NGT), and 551–553 (NGS) (H9 numbering), which differed from the 1998 vaccine strain, which had no glycosylation site at positions 551–553. Amino acid substitutions and variations were also observed in the NA protein sequences. The viral strains isolated in 2011 had lost the glycosylation site (NTT) at positions 70–72 and had the new glycosylation site (NSS) at positions 44–46 due to substitution of proline (P) to serine (S). Furthermore, one new glycosylation site (NSS) was present at positions 313–315 in A/Ck/Ir/189/2011, arising from a mutation of aspartic acid (D) to asparagine (N) (Table 2).
The amino acids of the hemadsorption (HB) site in the NA were examined at three locations within the molecule. Amino acids at positions 366–373 and 431–433 were IKKDSRAG and PQE, respectively. The HB site at positions 399–404 was DSDNLS in the vaccine strain, but DSDNRS in all the strains isolated in this study. There were no deletions in the stalk region of NA in any isolate.
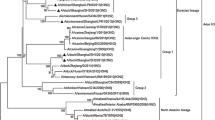
A phylogenetic analysis of the HA gene indicated that all the Iranian isolates belonged to the A/quail/Hong Kong/G1/97-like virus sublineage (Fig. 1a). The strains isolated in the present study were closely related to other Iranian strains that were dominant in 2009–2011. They were also closely related to H9N2 viruses from Pakistan, with identities of 94.3–97 %. The Iranian isolates clustered into three groups: (a) strains isolated in 1998–2005, (b) strains isolated in 2004–2009, and (c) strains isolated in 2010–2011, together with the new strains isolated in the present study (Fig. 1a). None of the sequences clustered within the Y280-like or Korean-like sublineage.

a Phylogenetic relationships between the HA genes from the H9N2 viruses isolated in Iran in 1998–2011 and other representative avian influenza viruses (AIVs) of this subtype. The strains isolated in this study are indicated by •. b Phylogenetic relationships between the NA genes from the H9N2 viruses isolated in Iran in 1998–2011 and other representatives AIVs of this subtype. The strains isolated in this study are indicated by filled circle. A Influenza A virus, Ck chicken, Ir Iran, Pak Pakistan, Av avian, S.Ara Saudi Arabia, Egy Egypt, Is Israel, Dub Dubai, HK Hong Kong, QA quail, Bej Beijing, Dk duck, Kor Korea
A phylogenetic analysis of the NA gene showed that all the NA genes of the Iranian H9N2 viruses fell into the G1-like sublineage (Fig. 2b). In turn, these Iranian viruses clustered into three groups: the first group included viruses isolated in 1998–2006, the second group included strains isolated in 2008–2009, and the third group comprised strains isolated in 2010–2011 and the currently circulating strains. Interestingly, the current strains and the strains isolated in Pakistan clustered in the same group, as they did in the HA gene analysis.

a Multiple alignment of HA amino acid sequences in the vaccine and field strains. b Multiple alignment of NA amino acid sequences in the vaccine and field strains
The H9N2 viruses included in the present study were compared antigenically with the vaccine strain. The chicken polyclonal hyperimmune serum raised against the vaccine strain (A/Ck/Ir/1221/98) showed fourfold reductions in cross-reactivity to the field viruses in the HI test (Table 1).
A comparison of the amino acid sequences of the HA proteins showed that five amino acid substitutions have occurred in the antigenic sites at positions D/N153G, L168S, S183D, T206N, and L234Q in the newly isolated viruses, and one amino acid substitution (V212I) has occurred in A/Ck/Ir/189/2011 (Fig. 2a).
Discussion
The phylogenetic analysis performed in this study demonstrates that the current Iranian and Pakistan isolates cluster in the same group, indicating that they might have originated from a common source. These results are consistent with those of previous studies that reported that recent isolates from Iran and Pakistan were very closely related and might have a common origin (Bashashati et al. 2013; Norouzian et al. 2014). In our phylogenetic analysis, the vaccine strain and the viruses isolated in 2011 clustered in different groups and displayed amino acid homologies in the HA and NA genes of 91.6–93.9 and 90.2–91.7 %, respectively. These results demonstrate that the currently circulating strains have nonnegligible genetic differences relative to the vaccine strain used in Iran. Identification of the antigenic sites of H9 is important for monitoring antigenic variants and for developing effective vaccines. Six amino acid substitutions, at positions 153, 168, 183, 206, 212, and 234, may reflect the antigenic drift observed in the 2011 field strains relative to the vaccine strain in Iran. It is remarkable that an inactivated H9N2 vaccine that has been used extensively to control AI might be responsible for the mutations observed in HA in the Iranian H9N2 viruses, resulting from the pressure on the chicken immune system exerted by the vaccination program. However, recent research suggests that selection by other factors, such as the specificity and avidity of the HA receptor and epistatic interactions within HA and between NA and other AI virus gene products, can select for changes in the globular region of HA, altering its antigenicity (Hensley et al. 2011; Kryazhimskiy et al. 2011). In this study, the HI test showed that these viruses are antigenically different from the vaccine strain. The field strains in the cross-HI studies showed fourfold reductions in inhibition, so the current vaccine seed strain might be ineffective (Li et al. 2005). For better protection, the match between the vaccine strain and the predominant circulating viruses must be maximized. Our results are consistent with those of other studies in which H9N2 viruses were phylogenetically characterized and their antigenic diversity was examined with the HI test (Park et al. 2011; Xue et al. 2014). In contrast, the H9N2 viruses isolated from chickens in Bangladesh in 2008–2011 were antigenically homogeneous. A phylogenetic analysis of the HA and NA genes of these isolates displayed monophyletic clusters with >95 % mutual homology (Shanmuganatham et al. 2013). Antigenic drift has been studied almost extensively in HA, although NA has also been shown to undergo antigenic drift (Sandbulte et al. 2011). NA-specific antibodies can reduce viral replication and disease severity in chickens (Webster et al. 1988). Despite this correlation with immunity, the antigenic drift of NA is not routinely examined. Sandbulte (2011) demonstrated that a single-point mutation in the NA protein was primarily responsible for the lack of inhibition exerted by a polyclonal antibody specific for earlier strains. These data emphasize the importance of antigenic drift when sequence changes occur in NA. The high variation in the amino acid sequences of NA (with a 9.8–8.3 % diversity) demonstrated in the present study may predispose the protein to antigenic change. It is recommended that an NI assay is used to antigenically characterize the NA protein (Sandbulte et al. 2011).
Analysis of the HA cleavage site showed that the strains isolated in 2011 contained a motif (KSSR/GLF) that differed from those previously reported in Iran in 1998–2008, when all the H9N2 viruses and the vaccine strain retained the conserved amino acid motif (RSSR/GLF) at the cleavage site (Homayounimehr et al. 2010; Moosakhani et al. 2010). The significance of this mutation for viral stability and pathogenicity warrants further study.
Glycosylation plays an important role in the viral life cycle. Glycans on the globular head of the HA can mask or modify antigenic sites recognized by neutralizing antibodies, thereby promoting virus survival in the face of vaccination or infection (Tate et al. 2014). In the present study, new glycosylation sites including NGS in HA and NSS in NA were detected in the currently circulating H9N2 strains. However, a more comprehensive analysis is required to determine whether they affect the antigenic sites on the virus.
Hemadsorption (HB) sites enhance the catalytic efficiency of NA. Changes in NA, as well as changes in the receptor-binding specificity of HA, are required for the emergence of pandemic influenza viruses (Uhlendorff et al. 2009). In this study, the amino acid arginine (R) was observed at residue 403 in the vaccine strain instead of leucine (L). The biological significance of this substitution in the HB site is unknown.
In summary, we have reported that the H9N2 influenza viruses currently circulating in Iran are closely related to strains isolated in Pakistan and might have originated from a common source. Our analyses reveal that the currently circulating strains and the vaccine strain are phylogenetically and antigenically distinguishable. These differences may reduce the efficacy of the commercially available vaccine in protecting the immunized hosts from infection with these new H9N2 viruses. We recommend surveillance of these new influenza strains and suggest that antigenic drift must be considered during the selection of influenza vaccine strains.
References
Alexander, D.J. and Spackman, D., 1981. Characterization of influenza A viruses isolated from turkeys during March-May 1979. Avian Pathology, 10, 281–293.
Bashashati, M., Vasfimarandi, M. and Sabouri, F., 2013. Genetic diversity of early (1998) and recent (2010) avian influenza H9N2 virus strains isolated from poultry in Iran. Archives of Virology, 153, 2089–2100.
Hall, TA., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hensley, S.E., Das, S.R., Gibbs, J.S., Bailey, A.L., Schmidt, L.M., Bennink, J.R. and Yewdell, J.W., 2011. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS ONE 6(2): e15190. doi: 10.1371/journal.pone.0015190.
Hoffmann, E., Stech, J., Guan, Y., Webster, R.G. and Perez, D.R., 2001. Universal primer set for the full-length amplification of all influenza A viruses. Archives of Virology, 146, 2275–2289
Homayounimehr, A.R., Dadras, H., Shoushtari, A. and Pourbakhsh, S.A., 2010. Sequence and phylogenetic analysis of the haemagglutinin genes of H9N2 avian influenza viruses isolated from commercial chickens in Iran. Tropical Animal Health and Production, 42, 1291–1297.
Kaverin, N.V., Rudneva, I.A., Ilyushina, N.A., Lipatov, A.S., Krauss S. and Webster, R.G., 2004. Structural differences among hemagglutinins of influenza A virus subtypes are reflected in their antigenic architecture: analysis of H9 escape mutants. Journal of Virology. 78, 240–249.
Kryazhimskiy, S., Dushoff, J., Bazykin, G.A. and Plotkin, J.B., 2011. Prevalence of epistasis in the evolution of influenza A surface proteins. PLoS Genetics, 7, e1001301. http://dx.doi.org/10.1371/journal.pgen.1001301.
Lee, D.H. and Song, C.S., 2013. H9N2 avian influenza virus in Korea evolution and vaccination. Clinical and Experimental Vaccine Research, 2, 26–33.
Li, C., Yu, K., Tian, G., Yu, D., Liu, L., Jing, B., Ping, J. and Chen, H., 2005. Evolution of H9N2 influenza viruses from domestic poultry in Mainland China. Virology, 340, 70–83.
Moosakhani, F., Shoshtari, A.H., Pourbakhsh, S.A., Keyvanfar, H. and Ghorbani, A., 2010. Phylogenetic analysis of the hemagglutinin genes of 12 H9N2 influenza viruses isolated from chickens in Iran from 2003 to 2005. Avian Diseases, 54, 870–874.
Nilli, H. and Assasi, K., 2003. Avian influenza (H9N2) outbreak in Iran. Avian Disease, 47, 828–831.
Norouzian, H., Bashashati, M. and Vasfimarandi, M., 2014. Phylogenetic analysis of neuraminidase gene of H9N2 avian influenza. Iranian Journal of Microbiology, 6, 91–97.
Okamatsu, M., Sakoda, Y., Kishida, N., Isoda, N. and Kida, H., 2008. Antigenic structure of the hemagglutinin of H9N2 influenza viruses. Archives of Virology, 153, 2189–2195.
Park, K.J., Kwon, H.I., Song, M.S., Pascua, P.N., Baek, Y.H., Lee, J.H., Jang, H.L., Lim, J.Y., Mo, I.P., Moon, H.J., Kim, C.J. and Choi, Y.K., 2011. Rapid evolution of low-pathogenic H9N2 avian influenza viruses following poultry vaccination programmes. The Journal of General Virology, 92, 36–50.
Sandbulte, M.R., Westgeest, K.B., Gao, J., Xu, X., Klimov, A.I., Russell, C.A., Bruke, D.F., Smith, D.J., Fouchier, R.A.M. and Eichelberger, M.C., 2011. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proceedings of the National Academy of Sciences, 108, 20748–20753.
Shanmuganatham, K., Feeroz, M.M., Jones-Engel, L., Smith, G.J., Fourment, M., Walker, D., McClenaghan, L., Alam, S.M., Hasan, M.K., Seiler, P., Franks, J., Danner, A., Barman, S., McKenzie, P., Krauss, S., Webby, R.J. and Webster, R.G., 2013. Antigenic and molecular characterization of avian influenza A (H9N2) viruses, Bangladesh. Emerging Infection Disease, 19, 1393–1402.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. 2011. MEGA5, Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution., 28(10), 2731–2739.
Tate, M.D., Job, E.R., Deng, Y.M., Gunalan, V., Maurer-Stroh, S. and Reading, P.C., 2014. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses, 6, 1294–1316.
Tong, S., Zhu, X., Li, Y., Shi, M., Zhang, J., Bourgeois, M., Yang, H., Chen, X., Recuenco, S., Gomez, J., Chen, L.M., Johnson, A., Tao, Y., Dreyfus, C., Yu, W., McBride, R., Carney, P.J., Gilbert, A.T., Chang, J., Guo, Z., Davis, C., Paulson, J.C., Stevens, J., Rupprecht, C.E., Holmes, E.C., Wilson, I.A. and Donis, R.O., 2013. New world bats harbor diverse influenza A viruses. PLoS Pathogens, 9(10):e1003657. doi: 10.1371/journal.ppat.1003657
Uhlendorff, J., Matrosovich, T., Klenk, H.D. and Matrosovich, M., 2009. Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Archives of Virology, 154, 945–957.
Vasfi Marandi, M. and Bozorgmehri Fard, M.H., 2002. Isolation of H9N2 subtypes of avian influenza viruses during an outbreak in chickens in Iran. Iranian Biomedical Journal, 61, 13–17.
Wan, Z., Ye J., Xu, L., Shao, H., Jin, W., Qian, K., Wan, H. and Qin, A., 2014. Antigenic mapping of the hemagglutinin of an H9N2 avian influenza virus reveals novel critical amino acid positions in antigenic sites. Journal of Virology, 88, 3898–3901
Webster, R.G., Reay, P.A. and Laver, W.G., 1988. Protection against lethal influenza with neuraminidase. Virology, 164, 230–237.
Xu, K.M., Li, K.S., Smith, G.J., Li, J.W., Tai, H., Zhang, J.X., Webster, R.G., Peiris, J.S., Chen, H. and Guan, Y., 2007. Evolution and molecular epidemiology of H9N2 influenza A viruses from quail in southern China, 2000 to 2005. Journal of Virology, 81, 2635–2645.
Xue, Y., Wang, J.L., Yan, Z.Q., Li, G.W., Chen, S.Y., Zhang, X.B., Qin, J.P., Li, H.Y., Chang, S.C., Chen, F., Bee, Y.Z. and Xie, Q.M, 2014. Sequence and phylogenetic analysis of surface protein genes of emerging H9N2 influenza viruses isolated from poultry in two geographical regions of China. Virus Genes, 48, 479–485.
Acknowledgments
We are grateful to the Department of Avian Disease and Research, at the Razi Vaccine and Serum Research Institute, Karaj, Iran, for the encouragement and the provision of facilities to carry out this work.
Authors’ contributions
The authors contributed equally to this work.
Conflict of interest
The authors declare that they have no competing interests.
Ethical standards
All animal studies were approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the Institutional Animal Care and Use Committee (IACUC) 1964.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bahari, P., Pourbakhsh, S.A., Shoushtari, H. et al. Molecular characterization of H9N2 avian influenza viruses isolated from vaccinated broiler chickens in northeast Iran. Trop Anim Health Prod 47, 1195–1201 (2015). https://doi.org/10.1007/s11250-015-0848-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-015-0848-x