
Abstract
Coronavirus disease-2019 (COVID-19) is a rapidly evolving health crisis caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). COVID-19 is a novel disease entity and we are in a learning phase with regards to the pathogenesis, disease manifestations, and therapeutics. In addition to the primary lung injury, many patients especially the ones with moderate to severe COVID-19 display evidence of endothelial damage, complement activation, which leads to a pro-coagulable state. While there are still missing links in our understanding, the interplay of endothelium, complement system activation, and immune response to the SARS-CoV-2 virus is a surprisingly major factor in COVID-19 pathogenesis. One could envision COVID-19 becoming a novel hematological syndrome. This review is to discuss the available literature with regards to the involvement of the complement system, and coagulation cascade and their interaction with endothelium.
Similar content being viewed by others
Highlights
-
SARS-CoV2 viral infection has been associated with a high incidence of venous thromboembolism and pulmonary embolism.
-
The pathophysiology and risk of thrombosis and embolism in patients with COVID-19 need elaborative research and discussion considering a higher morbidity and mortality risk.
Introduction
Currently, the World is facing an unparalleled public health challenge [1]. Since December 2019, the coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has relentlessly marched around the globe. Early reports from China revealed evidence of abnormal coagulation parameters associated with COVID-19 and its correlation to a more severe clinical course [2,3,4]. As the pandemic continues to spread in various parts of the World, frontline clinicians have quickly recognized an unusually high incidence of thromboembolic events in COVID-19 patients, including venous and arterial thrombosis [5]. Middeldorp et al. reported cumulated venous thromboembolism (VTE) incidences of 16%, 33%, and 42% in hospitalized COVID-19 patients on days 7, 14, 21, respectively, and a significant positive correlation between VTEs and disease severity and mortality [6]. Another multicenter analysis noted a significantly higher incidence of thrombotic complications in patients with COVID-19-related acute respiratory distress syndrome (ARDS) when compared to the matched non-COVID-19 ARDS population (11.7% vs. 2.1%, p < 0.008) [7]. So, both length and intensity of the infection and related illness play a role. For optimal management of COVID-19-associated coagulopathy (CAC), we need better understanding of the underlying pathophysiology [8].
Coagulation and COVID-19
Thromboinflammation
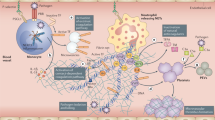
The concept of “thromboinflammation” means the activation of coagulation and thrombin generation as a result of systemic inflammatory responses induced by host defense system activation in the setting of viral, bacterial, or fungal infections and malignancies [9, 10]. The activation of coagulation in the setting of infection is a complicated multi-pathway process through extensive crosstalk pathways. When exposed to injury or infection, monocytes and subendothelial cells are activated and tissue factors are released to the blood, which triggers the generation of factor VIIa, factor Xa, and thrombin and plays a central role between the inflammation and coagulation pathways [11]. Proinflammatory cytokines and procoagulant microparticles are released by leukocytes, platelets and endothelial cells, promoting the upregulation of leukocytes adhesion molecules and suppression of vasculo-protective molecules [12]. These products ultimately promote the recruitment, adhesion and activation of platelets and leukocytes by forming neutrophil extracellular traps, which further recruit inflammatory leukocytes and promotes clotting and cytokine release [10, 12]. Disseminated intravascular coagulation (DIC) occurs when the localized adaptive hemostasis loses control and progresses into diffused microvasculature damage and subsequently, multiorgan failure [13].
It is important to note that although many COVID-19 patients do meet criteria for DIC or sepsis-induced coagulopathy (SIC) based on the International Society on Thrombosis and Haemostasis (ISTH) scoring systems [14], the coagulation parameters in COVID-19 differ from one seen in typical DIC [15]. The coagulopathy in COVID-19 presents with prominent elevation of d-dimer and fibrinogen degradation product (FDP) higher than what seen in SIC, while the hallmarks of decreased fibrinogen and profound thrombocytopenia in DIC are lacking [10, 15, 16]. Bleeding complications also appear to be uncommon in COVID-19 [15]. Increased level of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and IL-6 are associated with severe COVID-19 [17]. IL-6 is a potent driver for tissue factor expression. TNF-α and IL-1 further mediate the suppression of endogenous anticoagulant pathways [15]. Although not fully understood at the moment, the coagulopathy in COVID-19 appears to be linked to the extensive inflammatory responses leading to endothelial damage and significant activation of the fibrinolytic system [10, 15]. The coagulation proteins FXa and FIIa interact with SARS-CoV-2 Spike protein and induce conformal changes, which facilitate viral entry (infectivity). Hence anticoagulation especially during the early phase of viremia might inhibit SARS-CoV-2 replication [18].
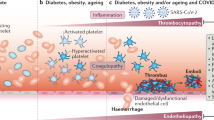
Endothelial dysfunction and thrombotic microangiopathy
SARS-CoV-2 uses angiotensin-converting enzyme-2 (ACE2) as a cell receptor to invade human cells. ACE2 is ubiquitously expressed in airway endothelial cells and also vascular endothelial cells [19]. Aside from the systemic inflammatory response, evidence also supports direct SARS-CoV-2 invasion to the vasculature. Monteil et al. demonstrated that SARS-CoV-2 could directly infect engineered human blood vessel organoids and human kidney organoids [20]. Autopsy of the lungs of severe COVID-19 patients showed severe endothelial injury associated with the presence of the intracellular virus and disrupted cell membranes [21]. Post-mortem analysis revealed viral inclusion structures, prominent endothelialitis, and apoptotic bodies in the vascular beds of several organs, including the kidney, heart, lung, and intestine [19]. The vascular endothelium is the principal organ in maintaining vascular tone and vascular homeostasis. The direct viral involvement and the profound host inflammatory response in COVID-19 cause endothelial dysfunction, which leads to inflammation with tissue edema and further triggers a prothrombotic state [22]. This correlates closely with the clinical observation that patients with chronic comorbidities which are associated with pre-existing endothelial dysfunctions, i.e. hypertension, diabetes, obesity, cardiovascular disease, chronic kidney disease, are at an increased risk for severe COVID-19 infection and adverse outcomes [23, 24].
In the setting of impaired microcirculation in vascular beds, diffused microvascular thrombi in the lung and multiple other organs have been described on autopsy review of COVID-19 non-survivors [21, 25]. The multisystem microvascular thrombosis, combined with laboratory abnormalities such as elevated lactate dehydrogenase (LDH), high ferritin concentration, low haptoglobin, mild anemia, elevated bilirubin level, schistocytes on peripheral blood smear and high prevalence of renal and cardiac injuries suggests that the coagulopathy in COVID-19 resembles thrombotic microangiopathy (TMA) [25,26,27].
TMA can occur in many different clinical scenarios, among which, transplant-associated TMA (TA-TMA) is a complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT) where a diffused endothelial injury causes microangiopathic hemolytic anemia and platelet consumption, resulting in microvascular thrombosis [28]. Evidence has suggested that high dose chemotherapy, radiation, calcineurin inhibitor exposure, graft-versus-host disease, and viral infections, i.e. BK virus and human herpesvirus 6, might be potential causative factors of TA-TMA [29, 30]. A relative ADAMTS13 deficiency [31], increased circulating endothelial cell (CEC) counts and serum levels of IL-6 and IL-8 [32] are observed in COVID-19, reminiscent the findings in TMA. Urokinase plasminogen activator receptor (uPAR) is bound on the endothelium and released into the blood stream as a soluble counterpart, named suPAR, in the setting of inflammation and endothelial damage [33]. Elevated suPAR level has been associated with severe COVID-19 and may serve as a prognostic factor for ARDS-associated with COVID-19 [33], which further supports the presence of endothelial injury and microangiopathy in COVID-19.
Complement activation in COVID-19
For any new infection, the initial immune response is provided by the innate immune system, which comprises of white cells (neutrophils, natural killer cells, macrophages, and dendritic cells) and complement. The innate immune responses are the first-line defensive mechanism against of pathogen invasion. They are also required to trigger the specific adaptive immune responses [34]. Complement cascade activation is therefore one of the earliest immune responses to infection [35, 36]. In early infection, pathogens prompt a local complement activation and inflammatory response. Depending upon the intensity of the initial injury and exposure over time, there might be over activation or dysregulation of the complement cascade, which could cause collateral damage of cells and tissues further leading to multiorgan dysfunction. Similar activation of complement cascade has been noted in the case of SARS-CoV-2 infection. The disseminated thrombotic phenomenon, cytokine release syndrome, and the overactivated complement system are amongst the major pathological mechanisms which in variable proportions are contributing towards worsening critical conditions of patients requiring intensive care support [37, 38]. This has been demonstrated in murine model studies conducted in SARS, and MERS [39, 40]. Gralinski et al. found that in the murine model lacking C3, the SARS-CoV infection severity, respiratory dysfunction, and cytokine levels were lower when compared to the ones with an intact complement system [1]. Similarly, in another murine model, Jiang et al. reported that blockade of C5 resulted in improved pulmonary functions, diminished cytokine response, and reduced viral load when compared no C5a blockage [39].
Also, data suggests that the pathological damage and organ dysfunction is likely due to the downstream effects of SARS-CoV-2 viral replication and further activation of immune pathways. Jiang et al. in their murine model on MERS infection noted a high concentration of C5a and soluble terminal complement complex activity (C5b-9) in sera and lung tissues suggesting the impact of excessive complement activation at the tissue level [39]. Similarly, Magro et al. did a histologic examination of the patients with COVID-19 and reported the presence of SARS-CoV-2 spike glycoproteins along with C4d and C5b-9 in the interalveolar septa and the cutaneous microvasculature [41]. The presence of complement activation products was detected on circulating erythrocytes in hospitalized patients with COVID-19 [42]. A recent preprint study reported that the highly pathogenic coronavirus N protein aggregated the lung injury caused by SARS-CoV-2, SARS-CoV, and MERS by binding to mannan-binding lectin-associated protease (MASP-2), a key serine protease that could directly activate the complement cascade. By blocking the N protein-MASP-2 interaction or suppressing complement activation, the lung injury could be significantly alleviated in vitro and in vivo [43]. Patients with severe COVID-19 treated with narsoplimab (anti-MASP-2 antibody) under compassionate use displayed rapid and sustained reduction in markers of endothelial damage (CECs, IL-6, IL-8, CRP and LDH levels). All patients treated with narsoplimab recovered and the agents is being evaluated further in patients with severe COVID-19 [32]. As mentioned above, the laboratory and autopsy findings in COVID-19 reminiscent of one seen in TMA, which is also highly associated with pathogenic complement activation [25]. Atypical hemolytic-uremic syndrome (aHUS) is one such rare immunological condition that is characterized by microangiopathy, hemolysis, low platelets, and acute renal failure. Amongst the infections, viral infections have also been studied as a potential trigger for TMA [44, 45]. Direct endothelial injury, cytokine storm-mediated endothelial injury, immune complex-mediated, complement dysfunction, and ADAMTS13 deficiency are amongst the major proposed mechanisms for the development of TTP/HUS type picture in viral diseases. Complement overactivation, measured as elevated C5b-9 in the blood, is a high-risk feature in TA-TMA and is associated with multiorgan injury and dismal outcomes [46]. Jodele et al. recently reported in the largest ever cohort series that terminal complement blockage with C5 inhibitor eculizumab significantly improved 1-yr post-allo-HSCT survival in patients with high-risk TA-TMA [46]. In COVID-19 disease, there is evidence suggesting complement dysregulation related TMA as one of the important pathophysiological mechanism in addition to or as part of thromboinflammation [25, 27, 47].
Recent data from Ramlall et al. observed that patients with macular degeneration, one of the most prevalent complement-mediated diseases resulting in enhanced complement activation, were at significantly higher risks for mechanical ventilation and mortality associated with COVID-19 [48]. Toll-like receptor 3 (TLR3) is one of the receptors of innate immunity that recognizes viral genomic RNA or the intermediates during viral replication, including double-stranded RNA (dsRNA). TLR3-knockout mice were more susceptible to SARS-CoV than wild-type mice but experienced only transient illness with no mortality [49]. Also, specific TLR3 polymorphism can be protective of age-related macular degeneration [50]. Evolving evidence indicates that extensive crosstalk exists between complement and TLR signaling pathways that may reinforce innate immune response and regulate inflammation [51]. Ramlall et al. also noted that none of the (only) four patients with complement deficiency in the whole cohort required ventilation or died after contracting SARS-CoV-2 [48]. Although exceedingly small in number, these findings are hypothesis-generating. Since patients with complement deficiency are typically considered severely immunocompromised, but in COVID complement deficiency or blockage might be protective.
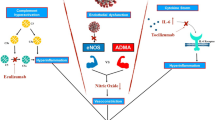
Role of complement inhibitors in COVID-19
With increasing evidence linking COVID-19, complement system and the resultant TMA together, and the emerging role of complement inhibition recognized in TA-TMA management, the next step for the research community is to test complement inhibitors in COVID-19 [46]. While multiple institutional experiences with investigational drugs such as tocilizumab, remdesivir, and convalescent plasma therapy in patients with COVID-19 have been reported [52,53,54,55], the data remain sparse with use of complement inhibitors. Eculizumab, an anti-C5 inhibitor, has shown some encouraging results in a small study in COVID-19 patients as recently reported by Diurno et al. [56]. Four critically ill patients with COVID-19 ARDS received injection eculizumab 900 mg for 2 doses, in addition to the other agents. There was a complete recovery in all four patients with a reduction in inflammatory markers.
Proximal complement inhibitors that target C3 or its upstream activators, which could theoretically provide broader complement inhibition, are currently under clinical development [57]. AMY-101, one of the next-generation C3 inhibitors, has been shown to interfere interleukin-6 release, a hallmark for CRS and severe COVID-19 infection, in ex vivo whole blood infection models [58, 59]. The first case report from Italy demonstrated successful compassionate use of AMY-101 in a patient with severe ARDS due to COVID-19 [60]. The limitations of these observational studies are the small patient population, lack of control arms, and concomitant use of other anti-COVID-19 drugs. SOLID-C19 (NCT04288713), CORIMUNO19-ECU (NCT04346797) and SAVE (NCT04395456) trials are amongst the few ongoing trials exploring the therapeutic effect and tolerance of complement inhibitors in patients with COVID-19. Medications targeting TLR signaling pathway, and thus its synergetic crosstalk with complement activation, maybe a potential treatment option as well [61].
Conclusion
Taken together, COVID-19-associated coagulopathy appears to be a complex pathological process caused by a cascade of thromboinflammation, endothelial injury, and complement activation, which results in a TMA. Current evidence supports that early use of corticosteroid [62], remdesivir [55] and/or convalescent plasma [52] is associated with improved outcome. The best therapy for critically ill COVID-19 patients who are beyond the reach of the three therapies is not known. Complement pathway inhibition may become the new target in treating critically ill patients in whom significant complement activation has occurred. One hopes that properly designed clinical trials will provide us with insight into the proper timing of complement inhibition, reliable clinical indicators to identify patients who would benefit the most from complement inhibition, and optimal usage of complement inhibition whether in a combination of anti-viral therapy or other anti-inflammatory or immune-based therapies that mitigate the cytokine storm in COVID-19.
Data availability
Author has seen the manuscript and agree to the content and data.
References
Sahu KK, Kumar R (2020) Current perspective on pandemic of COVID-19 in the United States. J Family Med Prim Care 9(4):1784–1791
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, Chine: a descriptive study. Lancet 395:507–513
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323(11):1061–1069
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan China: a retrospective cohort study. Lancet 395:1054–1062
Helms J, Tacquar C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46:1089–1098
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Muller MCA et al (2020) Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. https://doi.org/10.1111/jth.14888
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al (2020) High risk of thrombosis in patients in severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 4:1–10
Sahu KK, Siddiqui AD (2020) From hematologist’s desk: the effect of COVID-19 on the blood system. Am J Hematol 95(8):E213–E215
Sahu KK, Law AD, Kumar G, Dhir V, Naseem S, Nada R, Varma SC, Malhotra P (2016) Unusual cause of tophi with renal thrombotic microangiopathy. Indian J Hematol Blood Transfus 32(Suppl 1):100–103
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood 135(23):2033–2040
Butenas S, Orfeo T, Mann KG (2009) Tissue factor in coagulation: Which? Where? When? Arterioscler Thromb Vac Biol 29(12):1989–1996
Foley JH, Conway EM (2016) Cross talk pathways between coagulation and inflammation. Circ Res 118:1392–1408
Taylor FB, Toh CH, Hoots WK, Wada H, Levi M et al (2001) Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 86:1327–1330
Iba T, Levy JH, Warkentin TE, Thachil J, van der Poll T, Levi M et al (2019) Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Thromb Heemost 17:1989–1994
Levi M, Thachil J, Iba T, Levy JH (2020) Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol 7(6):e438–e440
Taylor FB, Toh CH, Hoots WK, Wada H, Levi M (2001) Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 86(5):1327–1330
Huang C, Wang Y, Li X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet 395:497–506
Lin L, Lu L, Cao W, Li T (2020) Hypothesis for potential pathogenesis of SARS-CoV-2 infection—a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect 9(1):727–732
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet Lond Engl 395(10234):1417–1418
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M et al (2020) Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181(4):905–913
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 383:120–128
Bonetti PO, Lerman LO, Lerman A (2003) Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol 23(2):168–175
Ghirga G (2020) Pre-existing endothelial dysfunction:*m a unifying hypothesis for the burden of severe SARS-CoV-2. BMJ 368:m627
Mishra AK, Lal A, Sahu KK, Sargent J (2020) Cardiovascular factors predicting poor outcome in COVID—19 patients. Cardiovasc Pathol 49:107246
Campbell CM, Kahwash R (2020) Will complement inhibition be the new target in treating COVID-19 related systemic thrombosis. Circulation 141:1739–1741
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet Lond Engl 395(10223):497–506
Mishra AK, Sahu KK, Lal A, Sargent J (2020) Patterns of heart Injury in COVID-19 and relation to outcome. J Med Virol. https://doi.org/10.1002/jmv.25847
Epperla N, Li A, Logan B, Fretham C, Chhabra S, Aljurf M et al (2020) Incidence, risk factors for and outcomes of transplant-associated thrombotic microangiopathy. Br J Haematol. https://doi.org/10.1111/bjh.16457
Dvorak CC, Higham C, Shimano KA (2019) Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Front Pediatr. https://doi.org/10.3389/fped.2019.00133
Haines HL, Laskin BL, Goebel J, Davies SM, Yin HJ, Lawrence J et al (2011) Blood, and not urine, BK viral load predicts renal outcome in children with hemorrhagic cystitis following hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 17:1512–1519
Martinelli N, Montagnana M, Pizzolo F et al (2020) A relative ADAMTS13 deficiency supports the presence of microangiopathy in COVID-19. Thromb Res 193:170–172
Rambaldi A, Gritti G, Mico MC et al (2020) Endothelial injury and thrombotic microagiopathy in COVID-19: treatment with the lectin-pathway inhibitor narsoplimab. Immunology. https://doi.org/10.1016/j.imbio.2020.152001
Rovina N, Akinosoglou K, Eugen-Olsen J et al (2020) Soluble urokinase plasminogen activator receptor (suPAR) as an early predictor of severe respiratory failure in patients with COVID-19 pneumonia. Crit Care 24:187
Alberts B, Johnson A, Lewis J et al (2002) Molecular biology of the cell, 4th edn. Garland Science, New York
Nasab MG, Saghazadeh A, Rezaei N (2020) SARS-CoV-2-A tough opponent for the immune system. Arch Med Res. https://doi.org/10.1016/j.arcmed.2020.05.020
Sahu KK, Mishra AK, Lal A (2020) COIVD-19 disease: tackling a pandemic in 21st century. QJM 113(7):519–520
Noris M, Benigni A, Remuzzi G (2020) The case of Complement activation in COVID-19 multiorgan impact. Kidney Int. https://doi.org/10.1016/j.kint.2020.05.013
Lal A, Mishra AK, Sahu KK (2020) CT chest findings in coronavirus disease-19 (COVID-19). J Formos Med Assoc 119(5):1000–1001
Jiang Y, Zhao G, Song N, Li P, Chen Y, Guo Y et al (2018) Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg Microbes Infect 7(1):77
Gralinski LE, Sheahan TP, Morrison TE, Menachery VD, Jensen K, Leist SR et al (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio 9(5):e01753
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J et al (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 220:1–13
Lam LM, Murphy SJ, Kuri-Cervantes L, Weisman AR, Ittner CAG, Reilly JP et al (2020) Erythrocytes reveal complement activation in patients with COVID-19. MedRxiv Prepr Serv Health Sci
Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H et al (2020) Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. Infect Dis. https://doi.org/10.1101/2020.03.29.20041962
Lopez C, Kim J, Pandey A, Huang T, DeLoughery TG (2020) Simultaneous onset of COVID-19 and autoimmune haemolytic anaemia. Br J Haematol 190:31–32
Hamad H, Sahu KK, Dunn S, Milla L, Caffery A, Islam N (2020) Rifampin Induced thrombotic thrombocytopenic purpura. Indian J Hematol Blood Transfus. https://doi.org/10.1007/s12288-019-01249-9
Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J et al (2014) Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood 124(4):645–653
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W et al (2020) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382(17):e38
Ramlall V, Thangaraj P, Meydan C et al (2020) Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat Med. https://doi.org/10.1038/s41591-020-1021-2
Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT et al (2015) Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6:e00638-15
Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, Gibbs D, Tong Z, Chen H, Constantine R, Yang X, Chen Y (2008) Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. https://doi.org/10.1056/nejmoa0802437
Hajishengallis G, Lambris JD (2016) More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev 274(1):233–244
Sahu KK, Jindal V, Siddiqui AD, Cerny J, Gerber JM (2020) Convalescent plasma therapy: a passive therapy for an aggressive COVID-19. J Med Virol. https://doi.org/10.1002/jmv.26047
Sahu KK, Mishra AK, Martin K, Chastain I (2020) COVID-19 and clinical mimics. Correct diagnosis is the key to appropriate therapy. Monaldi Arch Chest Dis Arch Monaldi Mal Torace 90(2):1296
Fu B, Xu X, Wei H (2020) Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med 18(1):164
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC et al (2020) Remdesivir for the treatment of covid-19—preliminary report. N Engl J Med 383:992–994
Diurno F, Numis FG, Porta G, Cirillo F, Maddaluno S, Ragozzino A et al (2020) Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci 24(7):4040–4047
Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F et al (2020) Complement as a target in COVID-19? Nat Rev Immunol 20(6):343–344
Mastellos DC, Ricklin D, Lambris JD (2019) Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov 18(9):707–729
Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X et al (2020) COVID-19 infection: the perspectives on immune responses. Cell Death Differ 27:1451–1454
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC et al (2020) The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol Orlando Fla 215:108450
Hajishengallis G, Lambris JD (2016) More than complementing Tolls: Complement-toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev 271(1):233–244
RECOVERY Collaborative Group (2020) Dexamethasone in hospitalized patients with Covid-19—preliminary report. N Engl J Med. https://doi.org/10.1056/NEJMoa2021436
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author has no conflicts of interest to declare.
Ethical approval
The article doesn’t contain the participation of any human being and animal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, X., Sahu, K.K. & Cerny, J. Coagulopathy, endothelial dysfunction, thrombotic microangiopathy and complement activation: potential role of complement system inhibition in COVID-19. J Thromb Thrombolysis 51, 657–662 (2021). https://doi.org/10.1007/s11239-020-02297-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-020-02297-z