
Abstract
The COVID-19 is still pandemic due to emerging of various variant of concern of SARS-CoV2. Hence, it is devastating the world, causing significant economic as well as social chaos. This needs great effort to search and develop effective alternatives along with vaccination. Therefore, to continue drug discovery endeavors, we used chalcone derivatives to find an effective drug candidate against SARS-CoV2. Chalcone is a common simple scaffold that exists in many diets as well as in traditional medicine. Natural as well as synthetic chalcones have shown numerous interesting biological activities and are also effective in fighting various diseases. Hence, various computational methods were applied to find out potential inhibitors of 3CLPro using a library of 3000 compounds of chalcones. Firstly, the screening by structure-based pharmacophore model yielded 84 hits that were subjected to molecular docking. The top 10 docked compounds were characterized for stability by using 100 ns molecular dynamic (MD) simulation approach. Further, the binding free energy calculation by MMPBSA showed that four compounds bind to 3CLPro enzyme with high affinity, i.e., − 87.962 (kJ/mol), − 66.125 (kJ/mol), − 59.589 (kJ/mol), and − 66.728 (kJ/mol), respectively. Since chalcone is a common simple scaffold that is present in many diets as well as in traditional medicine, we suggest that screened compounds may emerge as promising drug candidates for SARS-CoV-2. These compounds may be investigated in vitro to evaluate the efficacy against SARS-CoV-2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The severe behavior of severe acute respiratory syndrome corona virus-2 (SARS-CoV-2) and its variant of concern has led to a deplorable situation in the world. Therefore, COVID-19 is still a pandemic problem resulting in massacred millions of people. Despite the availability of several vaccines and mass vaccination, the management of COVID-19 is not very much successful in many countries. Moreover, with the emerging frequent mutations in SARS-CoV-2, it has become more difficult to find a successful cure. Several studies have revealed that SARS-CoV-2 acquires mutations in the S protein to increase its affinity for ACE2, resulting in increased levels of cytokines, TNF-, and NF-kB [1] and Omicron strain of SARS-CoV-2 has posed more burden forcing drug discovery for the future coming problem. Some specific mutations of SARS-CoV-2 have been associated with concerns about vaccination efficacy and natural infection–derived immunity to reinfection [2]. Therefore, the search for new drug candidates against COVID-19 is very necessary and urgent. Since it is quite difficult to predict how the worldwide landscape of SARS-CoV-2 vaccines will be shaped by availability and production capacity under the pressure of new SARS-CoV-2 mutants. Therefore, it is important to investigate new potential drug candidates against COVID-19.
Hence, keeping in mind the stated problem, in this study, we screened chalcone derivatives against 3CLPro enzyme of SARS-COV-2. Chalcones are also known as α, β-unsaturated ketones and have a common chemical scaffold 1,3-diaryl-2-propene-1-on (Fig. 1A). They can be obtained from natural sources and have a wide distribution in fruits, vegetables, and tea. Several chalcone-based compounds have been approved as therapeutic agents. For example, metochalcone was used as a choleretic drug, and sofalcone was previously used as mucoprotective and an antiulcer drug [3, 4]. Numerous studies have been done on the antiviral effects of synthetic as well as natural chalcones. Several reports suggest that chalcone possesses remarkable inhibitory activities against viruses like HIV [5], influenza A [6], rubella virus [7], and SARS-CoV [8].

A Basic structure of chalcones. B Cartoon representation of one protomer and dimeric 3CLPro-inhibitor complex
These chalcone compounds also inhibit the production of many cytokines such as TNF‐α, IL‐1β, IL‐6, and IκB kinases (IKKs) are one of the most essential regulators of the nuclear factor kappa B (NF-κB) pathway, which is known to be a central mediator of immunological responses and inflammation. Suppression of the NF-κB is considered a promising strategy for disease treatment. The chalcones exhibit NF-κB inhibitory activity by the covalent modification of the IKK proteins [9].
Since the 3CLPro enzyme plays an essential role in mediating the replication of SARS-COV-2 and has a low similarity of 3CLPro with human genes, it is a well-proven target for the development of drugs [10]. Recently, several studies have reported in silico identification of potential compounds against the main protease of SARS-CoV2 [1, 11,12,13,14]. 3CLPro is also required for the proteolytic release of enzymes important for viral replication, such as Nsp13, which has NTPase and RNA helicase activity [15]. It has been also proven that 3CLPro forms a homodimer (protomer A and protomer B) and that each of its monomers occupies 306 amino acids, including 3 domains, which fold into helices and β-strands. 3CLPro’s domains I (residues1–101) and II (residues 102–184) have an antiparallel-barrel structure, whereas domain III (residues 201–303) is made up of five helices grouped in a mainly antiparallel globular cluster and is linked to domain II via a lengthy loop region (residues 185–200).SARS-COV-2’s catalytic activity is mediated by a catalytic dyad (H41 and C145) located at the intersection of domain I and domain II (Fig. 1B), and S3, S2, S1, and S1′ are also significant subsites at the Mpro binding site [16].
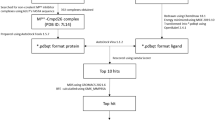
Recently, various studies have been performed to discover novel drugs against 3CLPro. In the present study, we aimed to test the potency of 3000 chalcone derivatives in inhibition of the target of SARS-CoV-2, 3CLPro through in silico methods. Therefore, structure-based virtual screening of the library is carried out with target protein, and molecular dynamic simulations were performed with the hit compounds. The drug-like properties and Lipinski’s rule were also predicted to ascertain the pharmacokinetics and drug ability of selected compounds.
Material and methods
Structure-based pharmacophore modeling and virtual screening
Library preparation
A library of 3000 compounds having chalcone scaffold was retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov) in SDF format and converted into PDB format by using Open Babel software.
Pharmacophore-based virtual screening
To determine the important pharmacophore features essential for the recognition of ligand by receptor and biological activity, structure-based pharmacophore modeling of SARS-CoV-2 3CLPro bound with its inhibitor X77 was carried out using Pharmit (http://pharmit.csb.pitt.edu/) webserver by loading the docked features. The model was constructed by using the selected PDB code 6W63 obtained from the RCSB protein data bank (https://www.rcsb.org/structure/6w63). The pharmacophoric model was generated by modifying the default parameters in the server. The generated model was utilized to identify potential lead compounds from the chalcone compound library. The hit compounds were arranged based on pharmacophore fit score and were used to further experimentation.
Molecular docking based virtual screening
Ligands and protein preparation
The generated output files or ligands from pharmacophore was further used for docking studies. Firstly, ligands were prepared for molecular docking analysis. The energy of the ligands was minimized using conjugate gradients optimization algorithm with a total number of 200 steps performed as a default universal force field (UFF) parameters [17]. The reference molecule X77 (Cid-145998279) was retrieved from the PubChem server. The docking requires the preparation of receptor protein by doing refinement processes such as addition and optimizing hydrogen bonds, removing atomic conflicts, and performing other operations. Hence, the crystal structure of 3CLPro (PDB ID: 6W63) protein was retrieved from the Protein Data Bank (https://www.rcsb.org). Further, all nonspecific molecules, water molecules, and ions were removed from the protein using PyMOL software. In addition, the hydrogen atoms were added to the protein receptor using the MGL Tools.
Molecular docking analysis
The molecular docking analysis was done with iGEMDOCK [18], a docking program that uses an empirical scoring function and the generic evolutionary method (GA).
The GA parameters are directly related to the docking performance. It has graphical user interfaces that detect pharmacological interactions and perform virtual screening. After generating a set of poses, iGEMDOCK recalculates the energy of each pose. The fitness is the total energy of the predicted pose in the binding site. The empirical scoring function of IGEMDOCK is estimated as follows:
Fitness = vdW (van der Waal) + Hbond (hydrogen bond) + Elec (electrostatic energy).
Using the specific docking function (slow docking), the ligand can be docked with the binding site of the protein. Finally, the iGEMDOCK analyzes and scores the screened molecule by combining pharmacological interactions with an energy-based rating system. The higher the negative energy scores, the stronger the binding affinity between the receptor and the ligands, whereas the positive energy value represents the poorer or no effective binding. Furthermore, the 2D and 3D interactions like hydrogen bonding and other non-bonded terms between the top docked compounds and the protein are seen using Accelrys Discovery Studio Visualizer software [19].
Molecular dynamic simulation
A molecular dynamic approach was conducted to assess the insight conformational changes and structural stability of 3CLPro and 3CLPro-ligand complex, by using Gromacs 5.0 package [20, 21]. Herein, the best poses obtained from re-docking studies, reference, and crystal structures of 3CLPro enzyme were subjected to MD simulation for 100 ns. The topology file of 3CLPro protein was generated by the GROMACS, while the ligand topologies were obtained from the CGenFF server. The simulations were performed using the CHARMM 36 force field [22], while the system was solvated with TIP3P water solvent model. Further, neutralization of all the complexes was done by the addition of ions. Energy minimization step was done for each system with the steepest descent algorithm using the Verlet cut-off scheme. In the next equilibrium phase, The NVT ensemble was done in 300 K with 5000 ps of steps. A total of 100 ns production simulations were performed with time step in a unit of 2 fs under NPT ensembles at a constant temperature of 300 K and 1.0 atm pressure using the Parrinello-Rahman for constant pressure simulation.
The generated trajectories were then used for the further calculations of free energy and analysis of various parameters like mean square deviation (RMSD), root-mean-square fluctuation (RMSF), and hydrogen bonds.
Binding free energy calculation using MM-PBSA
The inhibitory activity of selected compounds against target protein (3CLPro) predicted by the molecular docking and MD simulation studies can be validated by binding free energy calculations. Comprehensive analysis of the binding free energy (Gbind) of the selected ligands is performed using the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) approach implemented in the g_mmpbsa package[23]. The following equation describes the entire MM-PBSA process:
ΔGbinding = G complex − (G receptor + G ligand).
ΔGMM‐G/PBSA = ΔGvdw + ΔGele + ΔGpolar.
Finally, the gas-phase electrostatic energy (Eele), van der Waals (EvdW), polar (Gpolar), and nonpolar (Gnonpolar) components were added to calculate the MM-G/PBSA value of the protein–ligand complex.
In silico ADMET predictions
Evaluation of absorption, distribution, metabolism, and excretion (ADME) properties is an important criterion before a molecule is considered as a drug candidate. Hence, the freely accessible admetSAR ( http://www.admetexp.org) server was used to evaluate the ADME properties of top selected compounds.
Results and discussion
Pharmacophore-based virtual screening
Pharmacophore models were generated by using the structural and chemical similarity of the co-crystallized ligand. A pharmacophore is a set of spatial and electronic features required for interaction with a macromolecular target that results in a biological response. In this study, Pharmit server was used to construct a pharmacophore model. To obtain a pharmacophore model, 3CLPro complexed with X77 was used in Pharmit webserver.. A set of interacting pharmacophore characteristics from the 3CLPro-X77 complex was automatically generated by submitting the PDB ID of 3CLPro (6W63) to the Pharmit website which has been further modified.
The interactions between amino acids of 3CLPro-X77 were analyzed by DiscoveryStudio (see Fig. 2A). These interactions were considered for the selection of best pharmacophore model for screening purposes. In the selected model, the reference molecule (X77) showed three hydrogen bonds with HIS163, GLY143, and GLU166 and several hydrophobic bonds with MET165, ASN142, GLN189, HIS164, PRO168, HIS164, SER144, THR25, HIS172, THR26, PHE140, ASP187, and ARG188 residues of 3CLPro. X, Y, and Z coordinates for each feature are represented in Table 1. Thus, the selected model shows five features (Fig. 2B) that include one hydrogen bond acceptor for interacting with amino-acid residue GLY143, two hydrophobic centers, and two aromatic rings. Further screening with the above model resulted in 441 compounds. After that, compounds were minimized by mRMSD, and the AutoDockVina scoring function was used to compute the score. After minimization, 84 hits with a Max score of 0 and a Min RMSD value of 3 were obtained (Supplementary Table 1). These candidates were then downloaded as SDF format files from the Pharmit server and converted to PDB format for further analysis using Open Babel (version 2.3.1).

A Detailed view of 2D and 3D interactions of different amino acid residues within the binding pocket of COVID-19 3CLPro with the potent co-crystallized inhibitor X77 (PDB code: 6W63). B Manual generation of the pharmacophore as guided by the interactions of the potent inhibitor within the binding pocket of COVID-19 3CLPro
Molecular docking studies and interaction analysis
The generated pharmacophore-based 84 hits were further selected for the profound molecular docking studies by iGEMDOCK tool. The docking results were visualized and analyzed using Biovia Discovery Studio v4.1 visualizer. This enables the visualization of non-covalent interactions formed between ligand and protein, such as hydrogen bonds (classical and non-classical) and hydrophobic contacts (PI, alkyl). This helps to investigate the binding mode, affinity, and orientation of docked ligands at the active site of the receptor.
After the examination of the docked complexes, we investigated ten compounds having higher docking scores than the others (Table 2). As a result, these ten compounds were used for further investigation. The reference inhibitor (X77) with binding energy − 131.95 kcal/mol shows H-bond interactions with three amino acids within the binding pouch of 3CLPro such as GLU166, GLY143, and HIS41. Two pi-Alkyl bonds LEU27 and CYS145 were also observed with 3CLPro, while a pi-sulfur interaction MET49 was established between the aromatic rings. Two carbon-hydrogen bonds were formed with MET165 and ASN142 residues. Amide-pi stacked interactions emerged between LEU141. The remaining hydrophobic interactions (which are due to van der Waals forces) were formed with MET165, ASN142, GLN189, HIS164, PRO168, HIS164, SER144, THR25, HIS172, THR26, PHE140, ASP187, and ARG188 (Fig. 2A).
According to the above analysis, the compound 5035 shows a good bound energy of-147.76 kcal/mol. It also forms one hydrogen bond with ASP187, while MET49 and CYS44 establish two alkyl bonds. Residues HIS41, TYR54, ARG188, SER46, THR25, HIS164, LEU141, CYS145, ASN142, SER144, and HIS163 were associated with 3CLPro via van der Waals interactions.
Compound 44,405,163 with binding score − 143.39 kcal/mol binds within the 3CLPro active site through several non-covalent interactions. It binds to the active site through two conventional H-bonds with ASP187 and SER46, and one-carbon H-bond with MET49. 41 also found to be bound to active site through alkyl bond with MET49 and CYS44 residues (Fig. 3).

2D and 3D binding interaction between amino acid of top 10 compounds to the binding site of 3CLPro
Compound 101,389,938 (− 144.63 kcal/mol) stabilizes the 3CLPro active site through a hydrogen bond with PHE140 and via the formation of alkyl and Pi-alkyl bond with CYS145, HIS163, and MET165, and HIS141, respectively, and played an important role in the stabilization of the active site of the 3-chymotrypsin like protease. It formed hydrophobic interaction with residue ARG188, SER144, HIS164, and ASP187 via van der Waals forces.
Likewise, compound 41,145,803 with binding energy of − 145.08 kcal/mol also formed several non-covalent interactions with 3CLPro. However, it did not form any H-bonds with the protein 3CLPro. But it was dominated by hydrophobic interactions with 3CLPro through MET49, TYR54, GLN189, ASP187, CYS44, HIS164, ARG188, PRO52, MET165, GLU166, PHE140, SER144, LEU141, GLY143, and HIS172 residues. Meanwhile, residues CYS145 and HIS163 are found to bind with the 3CLPro through alkyl bond and, His41 formed Pi-cation interaction with 3CLPro.
Compound 11,784,736 had a van der Waals interaction with TYR237, LEU232, ALA234, VAL233, and LYS236 along with two conventional hydrogen bonds with ASN238 and ASN231. It shows relatively good binding affinity with a score of − 149.74 kcal/mol.
The binding energy of compound 10,648,096 with 3CLPro was − 148.73 kcal/mol. It interacted with ASP187, ARG188, GLN189, MET49, HIS163, PHE140, and LEU141 via van der Waals force (Fig. 3). Meanwhile, residues CYS145 and HIS41 are found to bind with the 3CLPro through a hydrogen bond and a carbon-hydrogen bond, respectively. It also formed an attractive charge with residue GLU166.
The result of compound 50,986,109 with a score − 146.85 kcal/mol indicated that GLU166 and HIS41 interact with the active site of 3CLPro by establishing hydrogen bonding interactions and pi-carbon bond, respectively. The hydrophobic residues THR25, THY54, ASN142, GLN189, ASP187, and GLY143 were found to strengthen the interactions between 3CLPro and compound 47 through van der Waals interactions.
In the case of compound 10,096,911, amino acid residues such as CYS145, GLY143, SER144, LEU141, GLU166, and HIS41 were identified as being associated with the formation of hydrogen bonds. Likewise, van der Waals hydrophobic interactions were established by residues THR190, GLN189, GLN192, LEU167, CYS44, HIS164, and ASN142. It also formed two carbon-hydrogen and pi-alkyl bonds between the protein and ligand. The binding energy of the compound is calculated as − 147.07 kcal/mol.
Compound 104,946 is stabilized within the active site of 3CLPro through binding energy − 143.82 kcal/mol and forms extensive van der Waals contacts with GLN189, THR190, ARG188, MET165, SER144, GLY143, ASN142, LEU141, and PHE140 residues. It was able to form one hydrogen bond and one carbon-hydrogen bond with residue CYS145 and HIS163, respectively. Meanwhile, it also showed one attractive charge with GLU166.
The compound 5,271,805 with a good binding score of − 160.61 kcal/mol forms two hydrogen bonds via MET165 and SER46 with 3CLPro. It also forms four carbon hydrogen bonds with residues ASP187, MET49, ARG188, and THR45. Likewise, hydrophobic interactions were established by residues PRO52, CYS145, HIS164, and TYR54 by van der Waals interactions. The residues involved in the binding of the top compounds are summarized in Table 3 and Fig. 3.
From these molecular docking results of the top protein–ligand complexes, we determined that all hit compounds have a similar binding affinity, which means that the screened compounds could potentially occupy the active site of the main protease very well, inhibiting the activity of 3CLPro to reduce the ability of virus replication. The common active side interacting residues between Mpro and hit compounds were Thr25, Leu27, His41, Cys44, His164, Asp187, Arg188, Cys145, Met49, and Met165. Finally, based on these observations, we finalize these 10 compounds for further analysis using MD simulations to investigate their stability and dynamic features.
Molecular dynamics and post-MM-PBSA analysis
The dynamic behavior and stability of the top 10 protein–ligand complexes were analyzed through 100 ns molecular dynamics simulation. All the trajectories were examined to understand the stability and the fluctuations of these complex structures through different parameters such as RMSD, RMSF, H-bond analysis, and MM-PBSA calculations. After RMSD analysis, out of ten compounds, four compounds, with CID (5035, 41,145,803, 44,405,163, and 101,389,938), showed promising inhibitory activity against 3CLPro. Therefore, only these four compounds were further analyzed and described.
Analysis of deviations in conformational elements
The calculation of root-mean-square deviation (RMSD) is the most significant and fundamental method for monitoring structural changes in protein–ligand complexes during molecular dynamics. It is the average displacement or deviations of a set of atoms for a particular frame. Protein with smaller deviations reflects a more stable nature [24]. RMSD of alpha carbon was analyzed for five systems. Plots for all the RMSD were generated by aligning the backbone structure of the complex formed during the MDS trajectory of 100 ns with the initial backbone structure (Fig. 4A). The average RMSD values for 3CLPro-X77 was 0.13 ± 0.01 nm, while for complexes, (5035-3CLPro), (41,145,803-3CLPro), (44,405,163-3CLPro), and (101,389,938-3CLPro) were found to be 0.14 ± 0.01 nm, 0.16 ± 0.01 nm, 0.14 ± 0.01 nm, and 0.15 ± 0.03 nm, respectively (Table 4). The RMSD of all the complexes was found to be stable and did not fluctuate. All the ligands revealed a minimal RMSD, which indicated that the protein–ligand complex was consistently maintained throughout the simulation period.

Various measures of the molecular dynamics simulations of top compounds with 3CLpro target: A RMSD, B RMSF, C Rg, D SASA, E projection on eigenvector, F interaction energy, G hydrogen number, and H eigenvalues
Fluctuations in the residual components
The method for identifying a flexible and rigid area in a protein is known as RMSF analysis. The greater the RMSF value reflects more flexibility and unstable the binding [25]. Variations in component residues were detected for protein-3CLPro and all complexes, during the 100 ns trajectory period. A very hydrophobic rich area forming loop exhibited a minimal change in the graph of 5035-3CLPro (Fig. 4B). Another tiny peak was discovered around residue 277, which was rich in hydrophobic residues once again. Residues 190–191 exhibited minimal variation in the case of 101,389,938-3CLPro (highly hydrophobic). All protein–ligand interactions had a fluctuation of less than 0.2 nm, which is completely acceptable (Table 4). The remaining complexes were found to be stable throughout the MD simulation period. These plotted data indicated that all the complexes showed active interaction with protein and remained stable during simulation.
Assessment of structural compactness
Rg provides insight into the system’s equilibrium conformation. It reflects a protein’s compactness. A low Rg denotes rigid packing, whereas a high Rg shows less flexibility. The binding of the ligands with protein can modify the conformation of the protein; therefore, with the help of Rg graphs, we can demonstrate that drug molecules have altered the folding behavior of the protein or retained it during MDS run [26]. The average Rg value for all the complexes was computed to be around 1.8 nm, and they remained in the plateau state excluding the complex (101,389,938-3CLPro) which was about 1.9 nm (Fig. 4C; Table 4). These findings indicate the strong binding of drugs to the active site of proteins as well as maintenance of compactness and stability of the 3d structure of the protein.
Intermolecular hydrogen bond analysis
Intermolecular hydrogen bonding is considered the most important parameter in drug discovery.
The binding of ligands is stabilized mainly by hydrogen bonds. The more the number of H-bonds, the higher the binding affinity toward protein [27]. The hydrogen bonds between each ligand–protein complex were investigated during 100 ns period (Fig. 4G). Many H-bonds are formed or deformed during MD simulations, but only a few of them are stable. These unstable H-bonds formed due to the motion of the protein and ligand atoms and may have a small contribution to the compound’s affinity toward 3CLPro. The maximum number of five H-bonds was formed in the case of 41,145,803-3CLPro. While in complexes XX7-3CLPro and 44,405,163-3CLPro, around 4 and 3 hydrogen bonds were computed, respectively. Similarly, around two hydrogen bonds were calculated for the complex 101,389,938-3CLPro and one for 5035-3CLPro. These observations suggest that 41,145,803 showed the highest bonding parameters followed by X77 and 44,405,163. Through the above data, it can be concluded that all the compounds are bound to 3CLPro in the same way as the reference (X77).
SASA and interaction energies analysis
The solvent accessible surface area (SASA) is a parameter calculated using the gmx_mmpbsa module of GROMACS. It measures the proportion of the interaction between complexes and solvents. SASA calculations can be used to estimate the extent of the conformational changes that occur during the binding process. The average SASA value of 149.2 nm2 was calculated for (X77-3CLPro) complex. In the case of complexes (5035-3CLPro) and (41,145,803-3CLPro), it was found to be around 149.9 nm2 and 150.9 nm2, respectively (Fig. 4D). Likewise, the complex (44,405,163-3CLPro) and (101,389,938-3CLPro) showed the average value of SASA to be around 148.3 nm2 and 151.2 nm2 (Fig. 4D). These calculations suggest that all the complexes were least exposed to the water solvent during 100 ns MD simulations which indicates the relatively stable nature of these complexes.
To confirm the binding scores obtained by molecular docking experiments, a comprehensive investigation was performed to quantify the free energy of interactions between protein–ligand complexes using the Parrinello–Rahman parameter implemented in GROMACS. In the 100-ns simulation period, the complex (444,405,163-3CLPro) was found to have the highest interaction energy of − 187.158 kJ/mol. Apart from this all the other complexes also showed good interaction energy which was significantly similar to that of the reference molecule (see Fig. 4F and Table 4).
Principal component analysis
PCA analysis was used to investigate the confined dynamical mechanical property, i.e., fluctuation and structural motion of all the complexes. The movements of the protein are usually determined by the some first eigenvectors [28]. It was observed, in this study, the first 40 eigenvectors accounted for the calculation of overall motion in each case.
The energetic participation of each complex for the motion was obtained by examining each eigenvector. The curves of eigenvalues (Fig. 4H) were produced after plotting eigenvalues against the eigenvectors. From the calculation, the motions for the first ten eigenvectors were accounted 74% for 3CLPro-X77, 74% for 3CLPro-101389938, 66% for 3CLPro-44405163, and 73% for 3CLPro-41145803 complex during 100 ns simulation period (Fig. 4H).
From the result, it can be suggested that all the complexes showed very fewer motions than the reference compound except complex 3CLPro-5035 which was calculated to be around 77%.
PCA can also be used to get the dynamics of the complexes by creating a 2D projection plot (Fig. 4E). In this investigation, we looked at the important movements of all the complexes using the first two principal components, PC1, and PC2.A complex that takes up less phase space and has a stable cluster is more stable, whereas a complex that takes up more space and has a non stable cluster is less stable. According to our findings, all of the complexes formed a very stable cluster and filled a smaller phase space.
In addition, the Gibbs energy graphs were also obtained from the PC1 and PC2 coordinates and are represented in Fig. 5. In these plots, ΔG values ranging from 0 to 12.5 kJ/mol, 0–13.5 kJ/mol, 0–13.7 kJ/mol, 0–11.8 kJ/mol, and 0–13.9 kJ/mol for 3CLPro-X77 complex, 3CLPro-5035 complex, 3CLPro-41145803 complex, 3CLPro-44405163 complex, and 3CLPro-101389938 complex, respectively. All the complexes represent significantly similar energy as the 3CLPro-X77 complex except the 3CLPro-44405163 complex which was slightly low. The result indicates that these compounds follow the energetically favorable transitions during the dynamics simulation.

Gibbs energy plot of reference and top 4 compounds complex with 3CLPro
Binding free energy calculations and energetic contribution of individual residues
The stability of the complex was further assessed by calculating the binding free energy of top compounds (last 20 ns) using the g_mmpbsa tool (Kumari et al., 2014). Complex3CLPro–101389938 was found to display the highest binding affinity or lowest binding free energy of -87.962 (kJ/mol).The ΔGBind of 3CLPro-5035, 3CLPro-41145803, and 3CLPro-44405163 complexes were found to be − 66.125 (kJ/mol), − 59.589 (kJ/mol), and − 66.728 (kJ/mol), respectively, which were better than the reference compound (− 61.700 kJ/mol). Overall binding of ligand and receptor is enhanced by energetic contributions from intermolecular electrostatic, van der Waals interactions, polar solvation energy, and the non-polar component of the free energy of solvation (SASA).The details of MM-PBSA calculation for the top four complexes are summarized in Table 5.
The MM-PBSA method was also used to create an energy profile to identify key residues involved in ligand binding to proteins. Figure 6 depicts the active site residues. According to the graph, the actively involved amino acid residues in all four compounds are His41, Thr25, Leu27, Met49, GLU166, Ser144, Cys145, Arg188, Met165, and Asp187. The per-residue interaction profile indicates that most residues have a negative binding affinity, which is crucial for the protein–ligand complex’s stability; however, some residues show a positive binding affinity. Thr25, Met49, Cys145, Asp187, and Met165 which have a negative binding affinity show a greater binding affinity and also play an important role in protein–ligand stability. The results of MD simulation and MM/PBSA analysis validate the molecular docking results. The high negative binding free energy of all four complexes demonstrates their stable configuration, which indicates that these compounds have enough affinity for 3CLPro to be considered as inhibitor drugs.

The contributions of individual amino acid residues of 3CLPro to the total binding during MMPBSA calculation
In silico ADME/pharmacokinetic predictions
ADME (absorption, distribution, metabolism, and excretion), which includes drug-likeness analysis, is important in drug development because it allows scientists to make appropriate decisions about whether inhibitors can be safely supplied to biological systems. In addition, inhibitors with poor ADME capabilities and severe toxic effects on biological systems are often the main reasons for the failure of drugs in clinical trials. Pfizer’s rule of five, also known as the Lipinski’s rule of five (5), is used to examine drug-likeness and to determine whether a specific inhibitor with biological and pharmacological characteristics would be an orally active drug in humans.
The rule states that a compound or an inhibitor can be active or orally absorbed if two (2) or more of these thresholds; molecular weight(Mw) of molecule < 500, octanol/water partition coefficient (iLOGP) ≤ 5, number of hydrogen bond acceptors(nHBA) ≤ 10, number of hydrogen bond donors (nHBD) ≤ 5, and topological polar surface area (TPSA) < 40 Å2) are not violated [29]. From the analysis, it was observed that all four compounds have zero or one violations, which is perfectly acceptable. Using admetSAR server, we extracted a few more data of top compounds. The outputs of some ADME and drug-likeness properties of the top 4 compounds are shown in Table 6. From the output, it was concluded that these compounds have lower BBB permeability than the reference compound. In the case of HIA analysis, it is well known that Greater HIA suggests that the compound could be better absorbed from the intestinal tract upon oral administration. If a compound with the HIA is less than 30%, it is labeled as HIA- otherwise; it is labeled as HIA + [30]. All selected compounds have above 30% HIA.
In terms of predicting the efflux by P-glycoprotein from the cell, all compounds come out to be non-inhibitor and substrate of P-glycoprotein except for compound 41,145,803 which was non-substrate. P-glycoprotein inhibitor indicates that the drug will inhibit the efflux process from the cell and increase bioavailability, whereas a non-inhibitor of P-glycoprotein means that the drug will efflux from the cell by the P-glycoprotein and pumped back into the lumen, limiting bioavailability and promoting the elimination of that drug in bile and urine [31].
In terms of metabolism, Cytochrome P450 monooxygenase (CYP) enzymes play important roles in drug metabolism have been extensively studied particularly 2D6, 2C9, and 3A4, which are most important in humans. For the CYP-2D6 and 2C9, we observed that all hit compounds were nonsubstrate (but non-inhibitor), whereas, in the case of CYP-3A4, reference molecule and compound 44,405,163 were shown to be metabolized by CYP450 since it comes out to be a substrate. For CYP-1A2, all the compounds were shown as a non-inhibitor (Table 6). A non-inhibitor of CYP450 refers that the molecule will not interfere with the biotransformation of drugs metabolized by CYP450 enzyme [32].
According to the carcinogenic profile, all the ligands were come out as non-carcinogenic similar to the reference molecule except compound 41,145,803. AMES toxicity reveals that all compounds were non-AMES toxic and can be used as a drug candidate.
The data of LD50 dose in rat model also computed by admetSAR server. The median lethal dose (PLD 50) is a specific measure of acute toxicity (dose that causes 50% mortality in treated animals when given over a period of time) that is used to compare the relative toxicity of various compounds. Acute toxicity refers to the negative effects of a compound that appear within a short time after exposure and is an important indication of drug safety in the early stages of toxicological research of unknown chemicals [30, 33]. By computing this property, we observed that all the compounds showed similar value as a reference molecule.
Overall, these ADMET and toxicity predictions suggest that some properties of compounds are associated with alerts, but a little modification in the structure can make these compounds more potent anti-COVID-19 drugs.
Conclusion
In our study, four chalcone compounds CID (5035, 41,145,803, 44,405,163, and 101,389,938) have been identified by the in silico approach, which may be able to inhibit the main protease of SARS-CoV-2. These selected compounds have a higher binding affinity ranging between − 143.39 and − 147.76 kcal/mol with 3CLPro. Initially, a structure-based pharmacophore model was developed using the 3D structure of 3-chymotrypsin like protease (3CLPro) and followed by molecular docking, MD simulation, and ADMET analysis. A total of 84 chalcone hits were captured by pharmacophore-based screening and were docked inside the active site of 3CLPro. The 10 compounds showed specific interactions within Mpro binding pocket that were comparable to reference X77. The MDS and MMPBSA analysis of four compounds gave promising results. These compounds were then refined using drug-like filters and ADMET analysis, which suggest that these compounds are non-carcinogenic and can be used as drug candidates. Taken together with the obtained data from this work and results, we suggest that the identified Chalcone candidates may serve as good scaffolds for the design of novel antiviral agents capable of targeting the active pocket of 3CLPro. The approach adopted here combining pharmacophore, docking, and molecular dynamics study is expected to facilitate the discovery of novel potential compounds against COVID-19 disease.
Data availability
All the data cited in this manuscript is generated by the authors and available upon request from the corresponding author.
Code availability
Not applicable.
References
Padhi AK, Tripathi T (2020) Can SARS-CoV-2 accumulate mutations in the S-protein to increase pathogenicity? ACS Pharmacol Transl Sci 3:1023–1026. https://doi.org/10.1021/acsptsci.0c00113
Collier DA, De Marco A, Ferreira IATM et al (2021) Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 593:136–141. https://doi.org/10.1038/s41586-021-03412-7
Opletalová V, Hartl J, Palát K, Patel A (2000) Conformational analysis of 2-hydroxy-2′,5′-diazachalcones. J Pharm Biomed Anal 23:55–59. https://doi.org/10.1016/S0731-7085(00)00263-6
Batovska D, Todorova I (2010) Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol 5:1–29. https://doi.org/10.2174/157488410790410579
Wu J-H, Wang X-H, Yi Y-H, Lee K-H (2003) Anti-AIDS agents 54. A potent anti-HIV chalcone and flavonoids from genus Desmos. Bioorg Med Chem Lett 13:1813–1815. https://doi.org/10.1016/S0960-894X(03)00197-5
Bizzarri BM, Fanelli A, Piccinino D et al (2019) Synthesis of stilbene and chalcone inhibitors of influenza A virus by SBA-15 supported Hoveyda-Grubbs metathesis. Catalysts 9:983. https://doi.org/10.3390/catal9120983
Ahmad A, Abdulrahman A AM (2008) The antiviral activity of the compound chalcone (4-ethoxy-2-hydroxy-4, 6-dimethoxy-chalcone) against rubella virus in vitro. Baghdad Sci J 5:391–394. https://doi.org/10.21123/bsj.5.3.391-394
Park J-Y, Ko J-A, Kim DW et al (2016) Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. J Enzyme Inhib Med Chem 31:23–30. https://doi.org/10.3109/14756366.2014.1003215
Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866. https://doi.org/10.1038/sj.onc.1203239
Wang M-Y, Zhao R, Gao L-J et al (2020) SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front Cell Infect Microbiol 10.https://doi.org/10.3389/fcimb.2020.587269
Nagar PR, Gajjar ND, Dhameliya TM (2021) In search of SARS CoV-2 replication inhibitors: virtual screening, molecular dynamics simulations and ADMET analysis. J Mol Struct 1246:131190. https://doi.org/10.1016/j.molstruc.2021.131190
Bojadzic D, Alcazar O, Chen J et al (2021) Small-molecule inhibitors of the coronavirus spike: ACE2 protein–protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect Dis 7:1519–1534. https://doi.org/10.1021/acsinfecdis.1c00070
Freitas FC, Ferreira PHB, Favaro DC, de Oliveira RJ (2021) Shedding light on the inhibitory mechanisms of SARS-CoV-1/CoV-2 spike proteins by ACE2-designed peptides. J Chem Inf Model 61:1226–1243. https://doi.org/10.1021/acs.jcim.0c01320
Gajjar ND, Dhameliya TM, Shah GB (2021) In search of RdRp and Mpro inhibitors against SARS CoV-2: molecular docking, molecular dynamic simulations and ADMET analysis. J Mol Struct 1239:130488. https://doi.org/10.1016/j.molstruc.2021.130488
Thiel V, Ivanov KA, Putics Á et al (2003) Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 84:2305–2315. https://doi.org/10.1099/vir.0.19424-0
Jin Z, Du X, Xu Y et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. https://doi.org/10.1038/s41586-020-2223-y
Rappe AK, Casewit CJ, Colwell KS et al (1992) UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc 114:10024–10035. https://doi.org/10.1021/ja00051a040
Hsu K-C, Chen Y-F, Lin S-R, Yang J-M (2011) iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics 12:S33. https://doi.org/10.1186/1471-2105-12-S1-S33
Stierand K, Maass PC, Rarey M (2006) Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics 22:1710–1716. https://doi.org/10.1093/bioinformatics/btl150
Pronk S, Páll S, Schulz R et al (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29:845–854. https://doi.org/10.1093/bioinformatics/btt055
Karplus M, McCammon JA (2002) Molecular dynamics simulations of biomolecules. Nat Struct Biol 9:646–652. https://doi.org/10.1038/nsb0902-646
Vanommeslaeghe K, Hatcher E, Acharya C et al (2009) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem NA-NA. https://doi.org/10.1002/jcc.21367
Kuzmanic A, Zagrovic B (2010) Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys J 98:861–871. https://doi.org/10.1016/j.bpj.2009.11.011
Sargsyan K, Grauffel C, Lim C (2017) How molecular size impacts RMSD applications in molecular dynamics simulations. J Chem Theory Comput 13:1518–1524. https://doi.org/10.1021/acs.jctc.7b00028
Papaleo E, Riccardi L, Villa C et al (2006) Flexibility and enzymatic cold-adaptation: a comparative molecular dynamics investigation of the elastase family. Biochim Biophys Acta - Proteins Proteomics 1764:1397–1406. https://doi.org/10.1016/j.bbapap.2006.06.005
Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42:623–628. https://doi.org/10.1134/S0026893308040195
Cyrański MK, Jezierska A, Klimentowska P et al (2008) Impact of intermolecular hydrogen bond on structural properties of phenylboronic acid: quantum chemical and X-ray study. J Phys Org Chem 21:472–482. https://doi.org/10.1002/poc.1389
Yang L-Q, Sang P, Tao Y et al (2014) Protein dynamics and motions in relation to their functions: several case studies and the underlying mechanisms. J Biomol Struct Dyn 32:372–393. https://doi.org/10.1080/07391102.2013.770372
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. https://doi.org/10.1016/S1056-8719(00)00107-6
Cheng F, Li W, Zhou Y et al (2012) admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105. https://doi.org/10.1021/ci300367a
Finch A, Pillans P (2014) P-glycoprotein and its role in drug-drug interactions. Aust Prescr 37:137–139. https://doi.org/10.18773/austprescr.2014.050
Cheng F, Yu Y, Shen J et al (2011) Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J Chem Inf Model 51:996–1011. https://doi.org/10.1021/ci200028n
Wang Y, Xing J, Xu Y et al (2015) In silico ADME/T modelling for rational drug design. Q Rev Biophys 48:488–515. https://doi.org/10.1017/S0033583515000190
Acknowledgements
The authors would like to thank the Head of the Botany Department, Soban Singh Jeena University, S.S.J Campus, Almora, India, for providing the requisite facilities to conduct this research work. Kumaun University, Nainital, is also acknowledged by the authors for providing high-speed Internet facilities. We also extend our appreciation to Rashtriya Uchchattar Shiksha Abhiyan (RUSA), Ministry of Human Resource Development, Government of India, for the deployment of computer infrastructure to establish Bioinformatics Centre in Kumaun University, S.S.J Campus, Almora.
Author information
Authors and Affiliations
Contributions
S. M. wrote the manuscript. T. J. has done all experiments parts. P. S. analyzed and interpreted data. S. C. conceptualized and designed the project. V. P. and S. C. supervised the study. The whole manuscript was approved by all authors.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• In silico screening of novel inhibitors against SARS-CoV-2.
• Ligand and structure-based drug development of chalcone compounds.
• Targeting 3CLPro to inhibit coronavirus using chalcones compounds.
• Molecular docking, MD simulation, and MM-PBSA studies of selected compounds.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mathpal, S., Joshi, T., Sharma, P. et al. Assessment of activity of chalcone compounds as inhibitors of 3-chymotrypsin like protease (3CLPro) of SARS-CoV-2: in silico study. Struct Chem 33, 1815–1831 (2022). https://doi.org/10.1007/s11224-022-01887-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-01887-2