
Abstract
COVID-19 is spreading in a global pandemic that is endangering human life and health. Therefore, there is an urgent need to target COVID-19 to find effective treatments for this emerging acute respiratory infection. Viral Papain-Like cysteine protease (PLpro), similar to papain and the cysteine deubiquitinase enzyme, has been a popular target for coronavirus inhibitors, as an indispensable enzyme in the process of coronavirus replication and infection of the host. Combined structure-based virtual screening, molecular dynamics (MD) simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) free energy calculation approaches were utilized for identification of PLpro inhibitors. Four compounds (F403_0159, F112_0109, G805_0497, D754_0006) with diverse chemical scaffolds were retrieved as hits based on docking score and clustering analysis. Molecular dynamics simulations indicated that the contribution of van der Waals interaction dominated the binding free energies of these compounds, which may be attributed to the hydrophobicity of active site of PLpro from COVID-19. Moreover, all four compounds formed conservative hydrogen bonds with the residues Asp164, Gln269, and Tyr273. We hoped that these four compounds might represent the promising chemical scaffolds for further development of novel PLpro inhibitors against COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since December 31, 2019, when the Chinese Center for Disease Control (China CDC) reported a cluster of severe pneumonia in the city of Wuhan in the Hubei Province, tens of millions of cases had been confirmed globally. It was found that the pathogen was a novel coronavirus previously unknown in humans, namely novel coronavirus 2019. On February 11, 2020, the World Health Organization (WHO) named it Corona Virus Disease 2019 (COVID-19) [1]. In light of the highly homologous between COVID-19 and severe acute respiratory syndrome (SARS), the International Virus Classification Commission (ICTV) classified the COVID-19 viruses as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [2, 3]. Common symptoms of COVID-19 infection included respiratory symptoms, fever, cough, and dyspnea. In more severe cases, the infection could lead to pneumonia, severe acute respiratory syndrome, kidney failure, and even death [4,5,6,7]. Unfortunately, to date, no specific therapeutic drug or vaccine had been approved for the treatment of COVID-19. Therefore, continuous efforts were urgent needed to find an effective treatment for this emerging acute respiratory infection.
Viral Papain-Like cysteine protease (PLpro, NSP3), similar to papain and the cysteine deubiquitinase (DUB) enzyme, had been a popular target for coronavirus inhibitors, as an indispensable enzyme in the process of coronavirus replication and infection of the host [8]. PLpro participated in the cutting and processing of the N-terminal of 1a(1ab) replicase protein and released the mature products nsp1, nsp2, and nsp3, which played an important role in regulatory protein molecule for the formation of SARS coronavirus replicase complex (RC) [9]. At the same time, PLpro had a negative regulating effect on the host’s antiviral innate immune response [10,11,12], and was an important interferon antagonist molecule of SARS coronavirus. Therefore, PLpro is an important target for the development of antiviral drugs. As one of the related targets of COVID-19, PLpro inhibitor research is also of great guiding significance for the targeted treatment of COVID-19 infection, but no inhibitor had yet been approved by the FDA for marketing.
In this study, combining structure-based virtual screening, molecular dynamics (MD) simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) binding free energy calculation were conducted on the crystal structure of PLpro to discover potent inhibitors against COVID-19 infection. Four compounds with diverse chemical scaffold were selected as hits, i.e., compounds F403_0159, F112_0109, G805_0497, and D754_0006. Furthermore, the binding affinities of PLpro inhibitors were evaluated by MM/GBSA binding free energy calculation and the detailed binding patterns were analyzed by MM/GBSA free energy decomposition.
Materials and methods
Structure-based virtual screening
Surflex molecular docking module in Sybyl-X2.1 (SYBYL_X2.1 is available from Tripos Associates Inc., S.H. R., St. Louis, MO, 631444, USA.) [13] was used for structure-based virtual screening. Crystal structure of the SARS-CoV-2 PLpro in complex with peptide inhibitor VIR251 (PDB ID: 6WX4) for virtual screening was obtained from the RCSB Protein Data Bank [14]. The ChemDiv database, a commercially available small molecule database from TopScience Co., Ltd. (Shanghai, China) containing more than 1.6 million compounds, was consulted as a screening library. Considering that only 2D structural information was available, all compounds in the ChemDiv database were preprocessed by using the db translate module in Sybyl-X2.1. During the preparation of protein PLpro, the space where ligand VIR251 was placed was selected as the inhibitor binding site, and all water molecules were removed. To accelerate the virtual screening, a high-speed screening was firstly carried out by decreasing maximum quantity of conformations and rotatable bonds from 20 to 10, and from 100 to 50, respectively. Then, the molecules with docking score within top 1% were screened again using the default docking parameters, which would result in 500 compounds.
To validate the accuracy of docking method and docking parameters, the molecular docking study was also applied to the crystal structure of SARS-CoV-2 PLpro complex with VIR251 with the same default docking parameters. Then, we compared the molecular conformations from docking with the one from crystal structure. After careful manual screening and clustering analysis, four compounds were hit. Moreover, to better investigate the binding affinities and binding modes of hit molecules, MD simulation and MM/GBSA free energy calculation were employed.
Molecular dynamics simulation
The initial conformations of PLpro-inhibitor complexes for MD simulation were obtained from the structure-based virtual screening. Four systems were modeled, i.e., PLpro-F403_0159, PLpro-F112_0109, PLpro-G805_0497, and PLpro-D754_0006. To eliminate bad inter-atom contacts or high energetic states of systems, all four systems were subjected to 20-ns MD simulation using the Amber 12 software package [15]. The parameter of protein PLpro was the standard Amber 03 force field (ff03), and those of the ligand inhibitors were the general Amber force field (GAFF). The inhibitors were minimized using the HF/6–31* optimization in Gaussian 09 program [16], and the partial charges were generated by fitting the electrostatic potentials derived by Gaussian program through the restrained electrostatic potential (RESP) fitting technique in Amber12. Each system was embedded in a truncated cathedral periodic box of TIP3P water molecules, which extends to at least 12 Å away from any solute atom. To neutralize the systems, appropriate numbers of sodium and chloride ions were added. Then, the water molecules and ions were primarily optimized with 2500 cycles of the steepest descent followed by 2500 cycles of conjugated gradient; the same minimization was next carried out by fixing backbone atoms; ultimately, energy minimization of 5000 cycles of the steepest descent and conjugated gradient were performed without any restriction to optimize the entire system. After energy minimization, each system was gradually heated in the NVT ensemble from 0 to 300 K within 60 ps. A 20-ns MD simulation with a time step of 2.0 fs was performed under a constant temperature of 300 K. The entire coordinate file was saved each 1 ps for the following binding free energy calculation.
MM/GBSA calculation and MM/GBSA free energy decomposition analysis
To calculate the binding free energies of the four PLpro-inhibitor complexes, MM/GBSA calculation was conducted via the following Eq. (1) as described in previous studies [17,18,19]:
where ΔEMM denoted the gas-phase interaction energies between ligands and proteins. It consisted of two parts, including van der Waals (ΔEvdw) and electrostatic (ΔEele) energies. ΔGGB and ΔGSA were polar and non-polar components of desolvation free energy, respectively; −TΔS was the entropy contribution at temperature T [20]. In this study, the polar solvation free energy was calculated by the Generalized Born (GB) model, with dielectric constants of the solvent and solute were set to 80 and 4, respectively. The non-polar contribution was estimated from the solvate-accessible surface area (SASA) by LCPO: ΔGSA = 0.0072 × ΔSASA. The binding free energy of each system was calculated based on 400 snapshots evenly extracted from 10- to 20-ns MD trajectories. Due to the high computational cost in the entropy calculation, only 100 snapshots were extracted from the last 10-ns MD trajectory which were used to calculate the entropic contribution.
To investigate the detailed interactions between protein PLpro and inhibitors, MM/GBSA free energy decomposition analysis was carried out via the mm_pbsa program in Amber 12 [21]. After the decomposition process, the energy contribution could be distributed to each residue of receptor and the binding interaction of each ligand-residue pair consists of three energy terms: van der Waals contribution (ΔEvdw), electrostatic contribution (ΔEele), and the desolvation term (ΔGdesolvation) which included the polar (ΔGGB) and the non-polar (ΔGSA) terms. All of the 400 snapshots generated for the binding free energies were also used for the free energy decomposition analysis.
Results and discussion
Structure-based virtual screening and binding modes of four hits
With the aim of testing and validating the Surflex docking-based virtual screening, molecular docking study was also employed to compare the conformations from docked structure with the one from crystal structure and the result is shown in Fig. S1. The superposition of these two conformations further demonstrated the accuracy of molecular docking method, which established the feasibility of the virtual screening in this study.
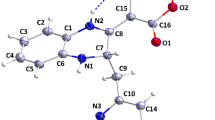
Four compounds (Fig. 1) were selected from the two-round virtual screening by ranking the docking scores and clustering analysis. The corresponding docking scores of four hit compounds including G805_0497, F112_0109, D754_0006, and F403_0159 were 10.93, 10.93, 10.62, and 10.61 (−log(Kd)), respectively. The scoring function was used to predict the binding affinities of protein/ligand complexes, with its output being represented in units of −log(Kd) [22, 23]. The higher docking scores implied the stronger binding affinities of these compounds to PLpro. The binding modes of four complexes are illustrated in Fig. 2. As shown in Fig. 2a and b, compound G805_0497 formed five hydrogen bonds with the residues Lys157, Leu162, Asp164, Gln269, and Tyr273. Meanwhile, it had strong hydrophobic interactions with the residues of binding site of PLpro including Leu162, Met208, Pro247, Pro248, Tyr268, and Try273. Similar to G805_0497, compounds F403_0159 (Fig. 2c and d), F112_0109 (Fig. 2e and f), and D754_0006 (Fig. 2g and h) also had hydrogen bond interactions with the residues Asp164, Gln269, and Tyr273. In addition, compound F112_0109 had one additional hydrogen bond with the residue Thr301, and compound D754_0006 formed π-π interaction with the residue Tyr268.

Structures of hit molecules from structure-based virtual screening

Predicted binding modes of four PLpro-inhibitor systems: PLpro-G805_0497 (a and b), PLpro-F403_0159 (c and d), PLpro-F112_0109 (e and f), and PLpro-D754_0006 (g and h) obtained from structure-based virtual screening. The protein PLpro is shown in cartoon and colored in blue. Hydrogen bond and π-π interactions are shown as dashed lines, and colored in red and blue, respectively
In light of the fact that the protein PLpro was treated as a rigid structure in the molecular docking–based virtual screening and the docking score might not represent the accuracy of binding affinity, combined MD simulation, and MM/GBSA free energy calculation which were applied on the four hit compounds.
The stability of MD simulation
Four PLpro-ligand complexes were conducted on 20-ns MD simulation. To explore the conformation dynamics of these four systems (PLpro-F403_0159, PLpro-F112_0109, PLpro-G805_0497, and PLpro-D754_0006), the root mean square deviation (RMSD) of backbone atoms of protein PLpro were calculated with the reference of the starting structure (Fig. 3). The RMSD plot of the whole protein indicated that the RMSD values of all four systems achieved equilibrium in a short time and fluctuated around 2 Å (Fig. 3), which suggested that the four compounds were quickly and steadily bound to the protein PLpro. In addition, the stationary value of each system was almost below 2 Å, which indicated that there existed strong binding interactions between receptor PLpro and four ligands. In light of the fact that all four systems were well equilibrated after 10 ns, so it is reasonable to perform the binding free energy calculation and free energy decomposition based on the last 10-ns trajectories.

RMSD of the protein backbone atoms (Cα, N, and C) of the four PLpro-ligand systems relative to the initial structures
In addition, the root mean square fluctuation (RMSF) of per amino acid residue of receptor PLpro was also calculated to evaluate the binding stability of PLpro with its inhibitors (Fig. 4). Overall, the RMSF distributions of these four models were similar, and the RMSF values of the key residues around the active site (i.e., Asp164, Arg166, Met208, Pro247, Pro248, Tyr264, Tyr268, Gln269, Tyr273, and Thr301) were lower than those in other regions of protein, which implied that these residues had strong binding interactions with the inhibitors. Moreover, RMSF calculation of each atom of four inhibitors was also carried out to evaluate the flexibility of all parts of inhibitor, and the results are depicted in Fig. S2 and Fig. S3. It is likely that the methyl and ethyl substituents in four inhibitors had higher RMSF, with the average RMSF values of 1.5 and 2.5 Å, respectively, while other parts of inhibitors fluctuated slightly. As shown in Fig. 2, the ethyl groups of compounds G805_0497 and F403_0159 were located outside the binding site and even exposed to the solvent.

Root mean square fluctuation (RMSF) of amino acid residue in four PLpro-ligand systems. Key amino acid residues for ligands binding are labeled and marked with purple dashed lines
Binding free energy calculated by MM/GBSA
MM/GBSA free energy calculation was employed to estimate the binding affinities of four ligands with protein PLpro, and the calculated binding free energies of four PLpro-ligand systems (PLpro-F403_0159, PLpro-F112_0109, PLpro-G805_0497, and PLpro-D754_0006) were − 26.59, − 19.96, − 15.76, and − 36.60 kcal/mol, respectively (Table 1). It is likely that compound G805_0497 exhibited the weakest binding affinity, while compound D754_0006 had the strongest binding interaction with PLpro. For all four systems, the van der Waals interactions (ΔEvdw) played a dominant role in the total binding free energy (ΔGbind). The contribution of electrostatic interaction (ΔEele) was nearly counteracted by the polar desolation free energy (ΔGGB).
Decomposition analysis of the binding free energies
MM/GBSA free energy decomposition analysis was employed to decompose the total binding free energies (ΔGbind) into ligand-residue pairs, which would provide more detailed information regarding the contribution of each residue for ligand binding (Fig. 5). It is obvious that the residue spectrograms of the four systems were similar to each other. The residues Asp164, Pro247, Pro248, Tyr264, Asn267, and Tyr268 played critical roles in compound G805_0497’s binding to protein PLpro; the free energy contributions of which were greater than 1 kcal/mol. The key residues for binding interaction between compound F112_0109 and PLpro were nearly the same as those in PLpro-G805_0497 systems.

MM/GBSA decomposition results of the total binding free energies per residue for four PLpro-ligand systems
Obviously, compared with the above two systems, compounds D754_0006 and F403_0159 had stronger binding affinities to the residues Asp164, Val165, and Arg166, which might result in the larger binding free energies of the two systems (Table 1). Moreover, compound D754_0006 had additional binding interaction with the residues Lys157, Leu162, and Gly163. Taken the results from free energy calculation and free energy decomposition analysis together, compounds D754_0006 and F403_0159 may have the outstanding binding modes with protein PLpro and might represent the new templates for designing potent PLpro inhibitors. It can be inferred that the residues Lys157, Leu162 to Glu167, Pro247, Pro248, Tyr264, Asn267 to Gln269, Tyr273, and Thr301 were the critical residues which dominated the binding interactions between PLpro and its inhibitors.
Conclusions
Considering that PLpro playing an important role in viral genome replication and escape from host innate immunity, we carried out a molecular modeling strategy via combining structure-based virtual screening, MD simulation, MM/GBSA binding free energy calculation, and free energy decomposition analysis to identify potent PLpro inhibitors for the treatment of COVID-19 infection. Four compounds (F403_0159, F112_0109, G805_0497, and D754_0006) with diverse chemical scaffolds were obtained from the structure-based virtual screening by targeting PLpro. MD simulations showed that the binding free energies of PLpro-D754_0006 and PLpro-F403_0159 systems were more potent than those of other systems, and the van der Waals interaction dominated the total binding free energies of PLpro-ligand systems. These two compounds had additional binding interactions with the residues Lys157 and Leu162 to Glu167 of PLpro, especially for compound D754_0006. It is likely that PLpro inhibitors would tightly bind to the residues Lys157, Leu162 to Glu167, Pro247, Pro248, Tyr264, Asn267 to Gln269, Tyr273, and Thr301. The present work would help in the further development of PLpro inhibitors against COVID-19, and give some valuable information on understanding the molecular mechanism of PLpro inhibitors.
References
Chen Y, Liu Q, Guo D (2020) Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 92:418–423
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506
Xie M, Chen Q (2020) Insight into 2019 novel coronavirus-an updated interim review and lessons from SARS-CoV and MERS-CoV. Int J Infect Dis 94:119–124
Wang W, Tang J, Wei F (2020) Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J Med Virol 92:441–447
Nascimento Junior JAC, Santos AM, Quintans-Júnior LJ, Walker CIB, Borges LP, Serafini MR (2020) SARS, MERS and SARS-CoV-2 (COVID-19) treatment: a patent review. Expert Opin Ther Pat:1–13
Rothan HA, Byrareddy SN (2020) The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun 109:102433
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, Ren R, Leung KSM, Lau EHY, Wong JY, Xing X, Xiang N, Wu Y, Li C, Chen Q, Li D, Liu T, Zhao J, Liu M, Tu W, Chen C, Jin L, Yang R, Wang Q, Zhou S, Wang R, Liu H, Luo Y, Liu Y, Shao G, Li H, Tao Z, Yang Y, Deng Z, Liu B, Ma Z, Zhang Y, Shi G, Lam TTY, Wu JT, Gao GF, Cowling BJ, Yang B, Leung GM, Feng Z (2020) Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. New Engl J Med 382:1199–1207
Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, El Oualid F, Huang TT, Bekes M, Drag M, Olsen SK (2020) Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv
Harcourt BH, Jukneliene D, Kanjanahaluethai A, Bechill J, Severson KM, Smith CM, Rota PA, Baker SC (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol 78:13600–13612
Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z (2014) SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein & Cell 5:369–381
Yuan L, Chen Z, Song S, Wang S, Tian C, Xing G, Chen X, Xiao ZX, He F, Zhang L (2015) p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J Biol Chem 290:3172–3182
Li SW, Wang CY, Jou YJ, Huang SH, Hsiao LH, Wan L, Lin YJ, Kung SH, Lin CW (2016) SARS coronavirus papain-like protease inhibits the TLR7 signaling pathway through removing Lys63-linked polyubiquitination of TRAF3 and TRAF6. Int J Mol Sci 17:678
SYBYL_X2.1 is availabe from Tripos Associates Inc. SHR, St. Louis, MO 631444, USA
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Case DA, Cheatham 3rd TE, Darden T, Gohlke H, Luo R, Merz Jr KM, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688
M. J. Frisch GWT, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016
An Y, Meng C, Chen Q, Gao J (2020) Discovery of small molecule sirt1 activator using high-throughput virtual screening, molecular dynamics simulation, molecular mechanics generalized born/surface area (MM/GBSA) calculation, and biological evaluation. Med Chem Res 29:255–261
Chen Q, Wang Y, Shi S, Li K, Zhang L, Gao J (2019) Insights into the interaction mechanisms of the proviral integration site of Moloney murine leukemia virus (Pim) kinases with Pan-Pim inhibitors PIM447 and AZD1208: a molecular dynamics simulation and MM/GBSA calculation study. Int J Mol Sci 20:5410
Wang E, Weng G, Sun H, Du H, Zhu F, Chen F, Wang Z, Hou T (2019) Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein–protein interactions. Phys Chem Chem Phys 21:18958–18969
Sun H, Duan L, Chen F, Liu H, Wang Z, Pan P, Zhu F, Zhang JZH, Hou T (2018) Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys Chem Chem Phys 20:14450–14460
Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, Hou T (2019) End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 119:9478–9508
Jain AN (1996) Scoring noncovalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des 10:427–440
Jain AN (2007) Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des 21:281–306
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 818 kb)
Rights and permissions
About this article

Cite this article
Pang, J., Gao, S., Sun, Z. et al. Discovery of small molecule PLpro inhibitor against COVID-19 using structure-based virtual screening, molecular dynamics simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) calculation. Struct Chem 32, 879–886 (2021). https://doi.org/10.1007/s11224-020-01665-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01665-y