
Abstract
A new series (6) of C5-curcuminoid derivatives (2E,6E-2,6-dibenzylidene-4-hydroxycyclohexanones) is described here with their evaluation for in vitro antiproliferative activities. Evaluation of 31 compounds against human A2780 (ovarian), C33A (cervix) and MDA-MB-231 (breast) cancer cell lines was performed to obtain structure activity relation data. The best performer was (2E,6E)-2,6-bis(3′-nitrobenzylidene)-4-hydroxycyclohexanone (6h) with IC50 values of 0.68 μM (A2780), 0.69 μM (C33A) and 0.92 μM (MDA-MB-231) compared to cisplatin with 1.30 μM, 3.69 μM and 19.13 μM, respectively. According to calculated physicochemical properties some members in series 6, namely (2E,6E)-2,6-bis[(4′-pyridinyl)methylene]-4-hydroxycyclohexanone (6p) [IC50 = 0.76 μM (A2780), 2.69 μM (C33A), 1.28 μM (MDA-MB-231)] seem to have improved bioavailability compared to curcumin. Selected members of series 6 were involved in circular dichroism spectroscopic measurements in order to determine their interaction with natural DNA. Based on these data, we conclude that these derivatives do not bind to DNA in vitro. A proposal is summarized based on mass spectrometric assessment for fingerprint analysis in biological research of such C5-curcuminoids.
Similar content being viewed by others
Introduction
Although there has been great progress in the development of treatment and prevention, cancer remains a major worldwide health problem. Because of an increase in worldwide mortality with growing prevalence, according to WHO reports [1], the successful treatment of cancer is still a challenge this century. Chemotherapy alongside surgical, radiological and biological treatment of cancer played a fundamental role for many decades. Hence, there is a current need to search for newer and more active anticancer compounds in pharmaceutical research fields. The research for mimics of natural materials is such a field in pharmaceutical science and is considered one of the major sources of lead molecules and drug candidates [2].
Plants used as spices, flavoring and coloring agents in the kitchen (nutraceuticals), or as preventive/curing additives in the Ayurvedic and folkloric medicinal practice (pharmaceuticals) are excellent sources for natural compounds. Such plants are common in the Zingiberaceae (ginger) family. Their powdered rhizome (a modified subterranean stem), as a yellow powder known as turmeric, has been used for thousands of years in Asian countries. For example, plants such as Curcuma longa L. and/or Curcuma domestica L. are also in use to prepare the well-known Indian spice turmeric currently. The major component of turmeric is its secondary metabolite, curcumin (1). Extensive research has shown that curcumin exhibits many different pharmacological effects and has a number of molecular targets with multiple pathways as an antiproliferative molecule [3, 4], although curcumin is remarkably non-toxic and has promising anticancer activities. Clinical studies indicate that its poor bioavailability and pharmacokinetic profiles due to instability under physiological conditions is a disadvantage [3]. Curcumin (diferuloylmethane), 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadien-3,5-dion contains a C7ß-diendione linkage (1,6-heptadien-3,5-dion) between the two arylidene groups in its structure. It was believed for a long time that natural curcuminoids (including the metabolites of curcumin) are exclusively C7-curcuminoids. However, along with curcumin a C5-curcuminoid was also isolated from both Curcuma longa and Curcuma domestica [5, 6]. This natural C5-curcuminoid, 1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadiene-3-one, as a truncated analogue of curcumin contains a C5ß-dienone linker (1,4-pentadiene-3-one) between the two benzylidene cores in its structure. This compound and its related derivatives proved to be more potent anticancer molecules compared to curcumin [7]. There is a great number of cyclic C5-curcuminoid analogues with excellent antiproliferative potential among the synthetic curcuminoids with the general structure 2 (Fig. 1). Structural modifications of curcumin and C5-curcuminoids focusing on enhancing their bioactivities have been investigated intensively during the last couple of decades. Numerous studies have been conducted, for example about the (3E,5E)-3,5-dibenzylidene-4-piperidone synthetic C5-curcuminoid family [8,9,10,11,12,13,14,15,16,17,18]. It is well known about these 4-piperidone derivatives that they exhibit higher cytotoxicity than curcumin towards different tumor cell lines such as breast, prostate, cervix, melanoma [3]. Compound EF24, for example (Fig. 1), showed high activity against cisplatin-resistant human ovarian cancer cells. The inhibitory effect of EF24 on cell-proliferation, as is common with such compounds, is due to its inductive ability to G2/M arrest and apoptosis by increasing phosphorylated phosphatase and tensin homolog (PTEN) expression [19]. In addition to G2/M arrest and apoptosis induction, these 4-piperidone derivatives show differential cytotoxicity: they are less toxic to non-cancerous cells when compared to cancer cells [8, 13,14,15,16].

Curcumin and synthetic cyclic C5-curcuminoids
Another crucial advantage of these synthetic C5-curcuminoids is that a number of them are able to revert multi-drug resistance (MDR) [17, 20, 21]. Moreover, there are reports on the in vivo tolerability and the lack of acute toxicity of these curcuminoids on rodents. There are compounds in this family that proved to be safe and did not cause mortality or convulsion in mice during a 2-week period of oral administration in 100–500 mg/kg dosage [22]. A number of C5-curcuminoids possessing the 1,4-pentadiene-3-one moiety have different modes of action, such as inducing apoptosis, cell cycle arrest, inhibiting the biosynthesis of polypeptides fundamental to tumor-progression, affecting mitochondrial respiration and stimulation/inhibition of certain enzymes playing role in tumor-growth [23]. Further, there is continuing debate on the binding or intercalation of curcumin onto DNA [24, 25] as to whether binding or intercalation would result in DNA damage in healthy cells as a dramatic side effect. There are scientific facts against this “DNA damage” theory in that curcumin binds to the minor groove of DNA, albeit weakly [26]. In another example, incubation with calf thymus DNA had little effect on either UV/Vis or fluorescence spectra of curcumin [27].
Based on the above description and the literature available in this field, we can say that the number of reports on the structural modifications on C5-curcuminoids is very high. So far, however, none of these drug-candidates passed into drug-development study or clinical trials.
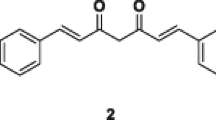
In view of the considerations above, we have decided to synthetize new, cyclic derivatives of natural C5-curcuminoids with the general structure 2 (Fig. 1). In one of our previous publications, we compared the antiproliferative activity of 25 arylidene cyclanones where the ketone scaffold was cyclopentanone, cyclohexanone, 1-indanone, 1-tetralone or 4-piperidone (2, X = CH2, NH or N-alkil) [28]. Their cytotoxicity against four human adherent cancer cell lines was evaluated. The cytotoxicity screen revealed that the diarylidene derivatives in general dominate the monoarylidene enones and, simultaneously, the nitrogen containing heterocycles display higher inhibition in cell-proliferation compared with the homocyclic analogs. The best performer was N-(γ-oxobutyl)-(3E,5E)-3,5-bis(4′-chlorobenzylidene)-4-piperidone (3, Fig. 1) with IC50 values of 0.438–1.048 μM. In the present article we describe the synthesis and comparative pharmacological evaluation of cyclic C5-curcuminoid analogues 2 (X = NCH3, CHCH3 or CHOH).
Experimental
Chemistry
Chemicals, solvents and reagents for the study were purchased from Alfa Aesar, Molar and Merck Ltd (Budapest, Hungary). Melting points were determined on a Barnstead-Electrothermal 9100 apparatus and are uncorrected. Silica gel 60 (0.2–0.5 mm, MERCK) was used for column chromatography and pre-coated silica gel 60 (F-254, MERCK) plates for TLC.
NMR: 1H- and 13C-NMR spectra were obtained using a Varian UNITY INOVA 400 WB spectrometer. Chemical shifts are referenced to the residual solvent signal. Measurements were run at a probe temperature of 298 K in CDCl3 or DMSO-d6 solutions. All the 1H- and 13C-NMR spectra were in accordance with the expected structures.
MS: The sample solutions were prepared by dissolving 0.2 mg synthesized molecules in 1000 µL acetonitrile/0.1% TFA (1:1, v/v). The clear sample solutions were pipetted onto the MALDI target plate (MTP 384 target plate, massive steel, Bruker Daltonics) After crystallization the mass spectra were acquired with an Autoflex II MALDI TOF/TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with a 337-nm pulsed nitrogen laser (MNL-205MC; LTB Lasertechnik Berlin, Berlin, Germany). MALDI measurements were performed in reflectron mode in a detection range of m/z 120 to 1000. Ions were accelerated under delayed extraction conditions (120 ns) in positive and in negative ion modes with an acceleration voltage of 20.00 kV. We used alpha-cyano-4-hydroxycinnamic acid (CHCA) matrix peaks as external calibration. The instrument was controlled and data was processed using FlexControl 2.4 and FlexAnalysis 3.4 software packages (Bruker Daltonics, Bremen, Germany.)
CD: CD and UV measurements were performed on a Jasco J-720 spectropolarimeter (Jasco Ltd., Tokyo, Japan) in Jasco cylindrical cuvettes with path length of 10 mm. One milligram of chicken erythrocyte DNA (Reanal, Budapest, Hungary) was dissolved in 10 mL distilled water (stock solution, 0.1 mg/mL) and was further diluted to 0.033 mg/mL for the experiments. Tested substances were dissolved in DMSO (Sigma-Aldrich, Budapest, Hungary) in 5–10 mM concentration. All experiments were performed at ambient temperature.
General method for the synthesis of compounds 6f, 6g, 6h, 6m, 6o, 6p (Method A: acid-catalyzed Claisen-Schmidt condensation)
A solution of 0.1 mol (1.141 g) 4-hydroxycyclohexanone and 0.2 mol of the appropriate arylaldehyde in 15 mL of glacial acetic acid was treated with 4 drops of concentrated sulfuric acid. This mixture was left to stand at room temperature until the starting materials couldn’t be detected on the TLC plate. The reaction mixture was then diluted with 100 mL water. After 10 min stirring the yellow solid was collected on a glass filter, treated with 25 ml of 5% aqueous solution of potassium hydrogencarbonate and washed with water. The yellow solid was dried and recrystallized from a suitable solvent.
(2E,6E)-2,6-Bis(4′-nitrobenzylidene)-4-hydroxycyclohexanone ( 6f )
Overall yield: 85%. Mp: 216–218 °C (MeOH/CHCl3); Lit.: 210–213 °C [42]. 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.06–3.18 (br m, 4H), 5.21 (m, 1H), 7.59 (d, J = 9.0 Hz, 4H), 7.92 (s, 2H), 8.29 (d, J = 9.0 Hz, 4H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 32.9, 66.9, 123.8, 130.7, 134.1, 137.4, 141.4, 147.6, 187.2. MS: m/z calculated for C20H16N2O6: see Table 1.
(2E,6E)-2,6-Bis(2′-nitrobenzylidene)-4-hydroxycyclohexanone ( 6g )
Overall yield: 81%. Mp: 178–179 °C (MeOH/CHCl3). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.84 (m, 2H), 2.95 (m, 2H), 4.97 (s, 1H), 7.54 (d, J = 7.5 Hz, 2H), 7.68 (t, J = 7.9 Hz, 2H), 7.83 (t, J = 7.5 Hz, 2H), 7.93 (s, 2H), 8.19 (d, J = 7.9 Hz, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 31.4, 67.2, 124.8, 129.8, 130.5, 130.9, 133.2, 133.8, 135.5, 147.7, 186.6. MS: m/z calculated for C20H16N2O6: see Table 1.
(2E,6E)-2,6-Bis(3′-nitrobenzylidene)-4-hydroxycyclohexanone ( 6h )
Overall yield: 79%. Mp: 168–169 °C (MeOH/CHCl3). 1H-NMR (500 MHz, acetone-D6) δ (ppm) 3.29 (m, 2H), 3.35 (m, 2H), 5.27 (m, 1H), 7.79 (t, J = 8.0 Hz, 2H), 7.90 (s, 2H), 7.99 (d, J = 8.0 Hz, 2H), 8.27 (d, J = 8.0 Hz, 2H), 8.36 (s, 2H). 13C-NMR (125 MHz, acetone-D6) δ (ppm) 34.2, 69.0, 125.2, 126.2, 126.6, 131.9, 136.4, 137.8, 137.9, 139.0, 188.6. MS: m/z calculated for C20H16N2O6: see Table 1.
(2E,6E)-2,6-Bis(4′-hydroxy-3′-methoxybenzylidene)-4-hydroxycyclohexano ne ( 6m )
Overall yield: 86%. Mp: 217–218 °C (MeOH). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.89 (m, 2H), 3.06 (m, 2H), 3.81 (s, 6H), 3.98 (s, 1H), 6.86 (m, 2H), 7.03 (m, 2H), 7.11 (s, 2H), 7.60 (s, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 36.6, 56.2, 64.2, 115.3, 116.0, 124.5, 127.4, 131.4, 137.8, 147.9, 148.3, 188.3. MS: m/z calculated for C22H22O6: see Table 1.
(2E,6E)-2,6-Bis[(3′-pyridinyl)methylene]-4-hydroxycyclohexanone ( 6o )
Overall yield: 77%. Mp: 198–199 °C (MeOH). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.94 (m, 2H), 2.98 (m, 2H), 4.08 (s, 1H), 4.97 (m, 1H), 7.49 (m, 2H), 7.68 (s, 2H), 7.96 (m, 2H), 8.57 (m, 2H), 8.74 (s, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 35.5, 62.8, 123.5, 131.1, 133.7, 135.5, 136.8, 149.2, 150.9, 187.8. MS: m/z calculated for C18H16N2O2: see Table 1.
(2E,6E)-2,6-Bis[(4′-pyridinyl)methylene]-4-hydroxycyclohexanone ( 6p )
Overall yield: 82%. Mp: 212–213 °C (MeOH). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.96 (m, 2H), 2.99 (m, 2H), 4.10 (m, 1H), 5.01 (m, 1H), 7.49 (d, J = 5.9 Hz, 4H), 7.59 (s, 2H), 8.65 (d, J = 5.9 Hz, 4H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 35.3, 39.4, 62.5, 124.0, 134.3, 137.2, 142.4, 149.8, 188.1. MS: m/z calculated for C18H16N2O2: see Table 1.
General method for the synthesis of compounds 6a, 6b, 6c, 6d, 6e, 6i, 6j, 6k, 6l, 6n, 6q (Method B: base-catalyzed Claisen-Schmidt condensation)
A solution of 0.1 mol (1.141 g) 4-hydroxycyclohexanone and 0.2 mol of the appropriate arylaldehyde in 20 mL of methanol was treated with a solution of 0.3 mol (1.683 g) potassium hydroxide in 20 mL of water. This mixture was stirred at room temperature on a magnetic stirrer until the starting materials couldn’t be detected on the TLC plate from the mother liquor. The reaction mixture was then diluted with 100 mL water. The precipitated yellow solid was collected on a glass filter and washed with water. The yellow solid was dried, and recrystallized from a suitable solvent.
(2E,6E)-2,6-Bis(benzylidene)-4-hydroxycyclohexanone ( 6a )
Overall yield: 88%. Mp: 139–140 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 1.94 (s, 1H), 2.98 (m, 2H), 3.19 (m, 2H), 4.12 (m, 1H), 7.45 (br m, 10H), 7.88 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.5, 65.6, 128.4, 128.8, 130.3, 132.3, 135.5, 139.3, 188.8. MS: m/z calculated for C20H18O2: see Table 1.
(2E,6E)-2,6-Bis(4′-chlorobenzylidene)-4-hydroxycyclohexanone ( 6b )
Overall yield: 86%. Mp: 167–168 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.97 (dd, J = 1.7, 7.8 Hz, 1H), 3.00 (dd, J = 1.7, 7.8 Hz, 1H), 3.14 (s, 1H), 3.17 (s, 1H), 4.18 (m, 1H), 7.38 (s, 8H), 7.82 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.4, 65.4, 128.8, 131.5, 132.6, 133.9, 135.0, 138.1, 188.3. MS: m/z calculated for C20H16Cl2O2: see Table 1.
(2E,6E)-2,6-Bis(2′,4′-dichlorobenzylidene)-4-hydroxycyclohexanone ( 6c )
Overall yield: 82%. Mp: 180–182 °C (MeOH/CHCl3). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.81 (m, 2H), 2.92 (m, 2H), 4.00 (m, 1H), 4.96 (s, 1H), 7.53 (br m, 4H), 7.71 (s, 2H), 7.76 (s, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 35.3, 63.1, 127.2, 129.1, 131.8, 132.3, 132.7, 133.9, 134.8, 136.0, 187.6. MS: m/z calculated for C20H14Cl4O2: see Table 1.
(2E,6E)-2,6-Bis(3′-chlorobenzylidene)-4-hydroxycyclohexanone ( 6d )
Overall yield: 85%. Mp: 120–122 °C (EtOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.99 (d, J = 7.6 Hz, 1H), 3.02 (d, J = 7.6 Hz, 1H), 3.14 (s, 1H), 3.17 (s, 1H), 4.19 (m, 1H), 7.32 (br m, 6H), 7.41 (s, 2H), 7.80 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.3, 65.3, 128.4, 128.9, 129.7, 129.8, 133.2, 134.4, 137.2, 138.0, 188.2. MS: m/z calculated for C20H16Cl2O2: see Table 1.
(2E,6E)-2,6-Bis(2′-chlorobenzylidene)-4-hydroxycyclohexanone ( 6e )
Overall yield: 80%. Mp: 152–153 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.85 (m, 2H), 3.02 (m, 2H), 4.11 (m, 1H), 7.26–7.32 (br m, 6H), 7.44 (m, 2H), 8.02 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.5, 65.9, 126.4, 129.8, 129.9, 130.3, 134.0, 134.1, 135.0, 136.7, 188.1. MS: m/z calculated for C20H16Cl2O2: see Table 1.
(2E,6E)-2,6-Bis(4′-dimethylaminobenzylidene)-4-hydroxycyclohexanone ( 6i )
Overall yield: 78%. Mp: 264–265 °C (MeOH/CHCl3). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.83 (m, 2H), 2.98 (s, 12H), 3.07 (m, 2H), 3.92 (s, 1H), 4.91 (s, 1H), 6.77 (d, J = 8.3 Hz, 4H), 7.41 (d, J = 8.3 Hz, 4H), 7.56 (s, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 36.5, 56.3, 64.0, 111.6, 122.9, 129.0, 132.0, 137.1, 150.3, 187.2. MS: m/z calculated for C24H28N2O2: see Table 1.
(2E,6E)-2,6-Bis(4′-methoxybenzylidene)-4-hydroxycyclohexanone ( 6j )
Overall yield: 81%. Mp: 162–163 °C (MeOH/CHCl3). 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.96 (dd J = 1.6, 8.4 Hz, 1H), 2.99 (dd J = 1.6, 8.4 Hz, 1H), 3.21 (d, J = 3.0 Hz, 1H), 3.24 (d, J = 3.0 Hz, 1H), 3.84 (s, 6H), 4.14 (m, 1H), 6.93 (d, J = 8.7 Hz, 4H), 7.44 (d, J = 8.7 Hz, 4H), 7.85 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.7, 55.3, 66.0, 114.0, 128.3, 130.4, 132.2, 138.9, 160.2, 188.6. MS: m/z calculated for C22H22O4: see Table 1.
(2E,6E)-2,6-Bis(3′,4′-dimethoxybenzylidene)-4-hydroxycyclohexanone ( 6 k )
Overall yield: 82%. Mp: 158–159 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm)2.99 (dd, J = 1.6, 8.2 Hz, 1H), 3.02 (dd, J = 1.6, 8.2 Hz, 1H), 3.21 (d, J = 2.4 Hz, 1H), 3.24 (d, J = 2.4 Hz, 1H), 3.90 (s, 6H), 3.91 (s, 6H), 4.17 (m, 1H), 6.92 (d, J = 8.4 Hz, 2H), 6.99 (d, J = 1.6 Hz, 2H), 7.09 (dd, J = 1.6, 8.4 Hz, 2H), 7.83 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.7, 55.9, 56.0, 65.8, 111.0, 113.8, 123.9, 128.5, 130.6, 139.2, 148.7, 149.9, 188.5. MS: m/z calculated for C24H26O6: see Table 1.
(2E,6E)-2,6-Bis(3′-methoxybenzylidene)-4-hydroxycyclohexanone ( 6 l )
Overall yield: 82%. Mp: 95–96 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.01 (m, 2H), 3.20 (m, 2H), 3.83 (s, 6H), 4.15 (m, 1H), 6.91 (d, J = 8.2 Hz, 2H), 6.97 (s, 2H), 7.04 (d, J = 8.2 Hz, 2H), 7.32 (m, 2H), 7.85 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 36.5, 55.3, 65.7, 114.5, 115.7, 122.7, 129.4, 132.6, 136.9, 139.3, 159.5, 188.7. MS: m/z calculated for C22H22O4: see Table 1.
(2E,6E)-2,6-Bis(4′-methylbenzylidene)-4-hydroxycyclohexanone ( 6n )
Overall yield: 86%. Mp: 155–156 °C (MeOH). 1H-NMR (500 MHz, DMSO-D6) δ (ppm) 2.34 (s, 6H), 2.91 (m, 2H), 3.05 (m, 2H), 3.99 (m, 1H), 4.93 (m, 1H), 7.28 (d, J = 8.0 Hz, 4H), 7.43 (d, J = 8.0 Hz, 4H), 7.63 (s, 2H). 13C-NMR (125 MHz, DMSO-D6) δ (ppm) 20.9, 35.9, 63.3, 129.1, 130.2, 132.5, 132.9, 138.8, 138.5, 188.1. MS: m/z calculated for C22H22O2: see Table 1.
(2E,6E)-2,6-Bis(cinnamylidene)-4-hydroxycyclohexanone ( 6q )
Overall yield: 87%. Mp: 195–196 °C (MeOH/CHCl3). 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.87 (m, 2H), 3.11 (m, 2H), 4.28 (m, 1H), 7.05 (br m, 4H), 7.31 (m, 2H), 7.36 (m, 4H), 7.50 (m, 4H), 7.56 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 35.2, 65.7, 123.3, 127.3, 128.8, 129.0, 131.6, 136.6, 138.6, 141.7, 187.3. MS: m/z calculated for C24H22O2: see Table 1.
General method for the synthesis of compounds 18a–d and 19
The appropriate bis(benzylidene)-cyclanone 17a–d or 6a (2 mmol) was dissolved in a warm (50–60 °C) solution of 35 mL dry toluene and triethylamine (2 mmol, 0.202 g). After removal of heating 2 mmol (0.226 g) chloroacetyl chloride in 10 ml of dry toluene was added dropwise to the above solution in 30 min under continuous stirring. Magnetic stirring was continued until the disappearance of the starting materials from the TLC plate. The mixture was evaporated to dryness under reduced pressure. The residue was triturated with 50 mL water and the precipitated yellow solid filtered off. After washing the solid several times with water the crystals were dried and recrystallized from a suitable solvent.
1-(α-Chloroacetyl)-(3E,5E)-3,5-bis(benzylidene)-piperidin-4-one ( 18a )
Prepared from (3E,5E)-3,5-bis(benzylidene)-piperidin-4-one (17a) [28]. Overall yield: 83%. Mp: 145–146 °C (toluene). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.88 (s, 2H), 4.78 (s, 2H), 4.91 (s, 2H), 7.40 (br m, 10H), 7.84 (s, 1H), 7.89 (s, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 40.6, 43.9, 46.7, 128.7, 128.9, 129.6, 129.7, 130.0, 130.5, 130.9, 134.2, 134.3, 137.8, 138.8, 165.2, 185.9. MS: m/z calculated for C21H18ClNO2: see Table 1.
1-(α-Chloroacetyl)-(3E,5E)-3,5-bis(4′-chlorobenzylidene)-piperidin-4-one ( 18b )
Prepared from (3E,5E)-3,5-bis(4′-chlorobenzylidene)-piperidin-4-one (17b) [28]. Overall yield: 81%. Mp: 169–170 °C (EtOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.91 (s, 2H), 4.77 (s, 2H), 4.88 (s, 2H), 7.28–7.48 (br m, 8H), 7.79 (s, 1H), 7.83 (s, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 40.6, 43.8, 46.9, 129.2, 129.3, 131.2, 131.3, 131.7, 132.7, 132.8, 136.0, 136.1, 136.8, 137.7, 165.2, 185.5. MS: m/z calculated for C21H16Cl3NO2: see Table 1.
1-(α-Chloroacetyl)-(3E,5E)-3,5-bis(4′-methoxybenzylidene)-piperidin-4-one ( 18c )
Prepared from (3E,5E)-3,5-bis(4′-methoxybenzylidene)-piperidin-4-one (17c) [28]. Overall yield: 80%. Mp: 162–163 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.82 (s, 6H), 3.92 (s, 2H), 4.75 (s, 2H), 4.88 (s, 2H), 6.93 (d, J = 7.9 Hz, 2H), 6.96 (d, J = 7.9 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 7,77 (s, 1H), 7.80 (s, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 40.9, 44.1, 47.0, 55.4, 114.4, 114.5, 127.0, 127.3, 128.9, 129.1, 132.2, 132.7, 137.4, 138.4, 160.9, 165.2, 185.8. MS: m/z calculated for C23H22ClNO4: see Table 1.
1-(α-Chloroacetyl)-(3E,5E)-3,5-bis(4′-dimethylaminobenzylidene)-piperidin-4-one ( 18d )
Prepared from (3E,5E)-3,5-bis(4′-dimethylaminobenzylidene)-piperidin-4-one (17d) [28]. Overall yield: 85%. Mp: 223–224 °C (EtOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.05 (m, 6H), 3.97 (s, 2H), 4.80 (s, 2H), 4.94 (s, 2H), 6.72 (m, 4H), 7.34 (m, 2H), 7.42 (m, 2H), 7.78 (s, 1H), 7.81 (s, 1H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 40.0, 41.0, 44.3, 47.1, 111.9, 122.3, 122.8, 126.6, 126.9, 132.4, 133.0, 137.7, 138.8, 151.0, 165.2, 185.7. MS: m/z calculated for C25H28ClN3O2: see Table 1.
(2E,6E)-2,6-Bis(benzylidene)-4-(α-chloroacetoxy)-cyclohexanone ( 19 )
Prepared from (2E,6E)-2,6-bis(benzylidene)-cyclohexan-4-one (6a). Overall yield: 85%. Mp: 159–160 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.22 (m, 4H), 3.97 (s, 2H), 5.31 (m, 1H), 7.35–7.47 (br m, 10 H), 7.94 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 32.8, 40.8, 69.7, 128.6, 129.1, 130.2, 130.8, 135.2, 140.0, 166.6, 187.8. MS: m/z calculated for C22H19ClO3: see Table 1.
General method for the synthesis of compounds 15 and 16
Oxaloyl chloride (2.5 mmol) was added to a stirred suspension of 5 mmol of the corresponding bis(benzylidene)cyclanone and 50 mL of dry toluene containing 5 mmol triethylamine at roughly 20 °C. The reaction mixture was stirred and left overnight at room temperature. Then this the mixture was diluted with 50 mL of water. The precipitated yellow solid was collected on a glass filter, washed thoroughly with water, dried and recrystallized from methanol.
1,2-Bis[(2E,6E)-2,6-bis(benzylidene)-4-oxycyclohexanone-4-yl]ethane-1,2-dione ( 15 )
Prepared from compound 6a. Overall yield: 85%. Mp: 247–248 °C (MeOH). 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.17 (m, 4H), 3.30 (m, 4H), 5.24 (s, 2H), 7.30–7.49 (br m, 20H), 7.93 (s, 4H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 32.7, 71.4, 128.6, 129.1, 130.2, 130.5, 135.1, 140.2, 156.6, 187.4. MS: m/z calculated for C42H34O6: see Table 1.
1,2-Bis[(3E,5E)-3,5-bis(benzylidene)-4-piperidon-1-yl]ethane-1,2-dione ( 16 )
Prepared from (3E,5E)-3,5-bis(benzylidene)-piperidin-4-one (17a) [28]. Overall yield: 88%. Mp: 243–244 °C; Lit.: 245–246 °C [18].
2,4-Dimethyl-1,5-diphenylpenta-1,4-dien-3-one (2e) ( 4 )
This compound was prepared according to a known method [45]. Overall yield: 40%. Mp: 125–127 °C; Lit.: 127–128 °C [45].
1-Methyl-(3E,5E)-3,5-bis(benzylidene)-4-piperidone (5a) and 1-Methyl-(3E,5E)-3,5-bis(4′-chlorobenzylidene)-4-piperidone ( 5b )
These compounds were prepared according to a known method [28]. Mp: 5a – 116–117 °C; Lit.: 116–117 °C [28]. 5b – 183–184 °C; Lit.: 184–185 °C [28].
(2E,6E)-2,6-Bis(benzylidene)-4-methylcyclohexanone (7a) and (2E,6E)-2,6-Bis(4′-chlorobenzylidene)-4-methylcyclohexanone ( 7b )
These compounds were prepared according to a known methods [46] and [47], respectively. Mp: 7a – 98–99 °C; Lit.: 98–99 °C [46]. 7b – 163–164 °C; Lit.: 156–160 °C [47].
Determination of antiproliferative action in vitro
The antiproliferative properties of the synthesized compounds were determined against a panel of human adherent cancer cell lines. Briefly, MDA-MB-231 (breast), C33A (cervical) and A2780 (ovarian) cancer cell lines cell were purchased from European Collection of Cell Cultures (ECCAC, Salisbury, UK) and maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids and an antibiotic–antimycotic mixture (Lonza Group Ltd., Basel, Switzerland). Near-confluent cancer cells were seeded onto a 96-well microplate (5000/well) and after overnight standing new medium, containing the tested compounds at 0.3, 3 and 30 µM, was added. After incubation for 72 h at 37 °C in humidified air with 5% CO2, the viability of the cells were determined by the addition of 20 µL of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution. The mitochondrial reductase of the intact cells metabolized the MTT and the produced formazan was precipitated as purple crystals during a 4-h contact period. The formazan was dissolved in 100 µL of DMSO during a 60-min period of shaking at 37 °C assayed at 545 nm, using a microplate reader [50]. In the case of the most active compounds the assays were repeated with a set of dilutions (0.03–10 mM), concentration–response curves were fitted to the generated data and the IC50 values were calculated by means of GraphPad Prism 4.0 (GraphPad Software, San Diego, CA, USA). The experiments were carried out on two microplates with five parallel wells. Stock solutions of the tested compounds (10 mM) were prepared in DMSO. The highest DMSO content of the medium (0.3%) did not have any substantial effect on the cell proliferation. Cisplatin (Ebewe Pharma GmbH, Unterach, Austria) was used as a reference agent.
Physiscochemical calculations
For the prediction of some physicochemical parameters of our compounds we have used the Calculator Plugins of ChemAxon. The ClogP values were calculated with the Klopman method and database of the software. The ClogP, pKa and solubility values are results at physiological pH = 7.4. See the corresponding homepage for detailed conditions of the calculations [43]. To explain the relationship between biodata and calculated physicochemical parameters the Lipinski approaches have been used [44].
Results and discussion
Design and synthesis
Our purpose is to give further insight into the influence of structural modifications at position 4 of the cyclic ketone scaffold on the biological effect. We proved among other things [28] that this position, being an auxiliary binding ability (secondary pharmacophore) to the in vivo biological site of action, has great influence on the cytotoxicity of these compounds. In other words, the binding strength of the C5-curcuminoids to the biological site of action is weak when merely the pentadienone moiety reacts. The substituent at position 4 on the cyclanone ring is able to enhance the interaction between the C5-curcuminoids and their biological place of action in the cancer cell. Therefore, we set out to synthesize compounds 4–7 for comparative structure–activity relationship (Fig. 2) investigation.

Acyclic (4) and cyclic (5–7) C5-curcuminoids
Syntheses of compounds in series 5–7 and 15–19 are outlined in Schemes 1, 2, 3 and 4. Briefly, compound 4 and the bis(arylidene)-cyclanones 5–7 were synthesized via acid- or base-catalyzed condensation of diethylketone, 4-hydroxycyclohexanone, 4-methylcyclohexanone or 4-piperidone hydrochloride with aryl aldehydes according to the Claisen-Schmidt condensation protocol [28].

The synthesis of series 6 from 4-hydroxycyclohexanone and aromatic aldehydes. Compound 12 was prepared according to a known method [38]

The synthesis of derivatives 6o-q

The synthesis of the diester derivative 15

The synthesis of chloroacetyl derivatives 18 and 19
Another objective was the synthesis of a new branch of cyclic C5-curcuminoid with a hydroxyl functionality at position 4 (6) and derivatives of series 6 with different aryl substituents (Schemes 1,2, 4.) to receive data on the impact of the corresponding structural changes on cytotoxicity.
The optimal para substituents (H, Cl, OCH3, NO2, OH, dimethylamino) on the arylidene moiety was selected according to a previous disclosure on this substituent effect [29]. “Optimal” means that the chosen substituents on the arylidene group have different electronic, hydrophobic and steric properties fitting well to Craig plot for para substituents assuring the divergence of the σp and π values [30, 31]. Special aromatic aldehydes like pyridincarbaldehyde and cinnamaldehyde were also used (Scheme 2).
The synthesis of the dimeric compound 15 was performed by acylation of the monomer 6a with oxaloyl chloride in the presence of triethylamine analogously to compound 16, which was prepared by others earlier [18] (Scheme 3).
Chloroacetyl derivatives 18 and 19 were obtained from their corresponding dibenzylidene precursors 6a and 17a–d with chloroacetyl chloride in the presence of trimethylamine (Scheme 4).
Structural characterization of the title compounds
Structures of the compounds synthesized were established by means of 1H, 13C NMR and MS measurements. The NMR spectra of compounds in series 6 were similar and simple in appearance. The E,E-configuration of the newly synthesized compounds was determined by 1H NMR [32]. The E,E-configuration is due to the anisotropic effect of the carbonyl group on the vinyl protons, which is not possible in the Z,Z-configurational forms.
Our research group is using, among others, matrix-assisted laser desorption ionization (MALDI) technique for single-stage MS analysis. Because of its large mass range, the device is a time-of-flight spectrometer (MALDI-TOF). This technique was used also in our previous study on cyclic C5-curcuminoids [28]. We have investigated a number of analytes in the series of 3,5-dibenzylidene-4-piperidones and 2,6-dibenzylidene-1-cyclanones. All compounds showed characteristic molecular ion peaks (Table 1). In general we can state that the diagnostic product ions were not only fragments, but also dimers or sometimes trimers of the molecular ions in their MS spectra. The dimerization is a common photochemical [2 + 2] cycloaddition. The photodimerisation, as a type of autodimerization, is well-known in this family of compounds [33,34,35,36]. The reaction proceeds via a stepwise sequence. The product is a dispiro cyclobutane derivative in general, as is compound 20 in our case. We have selected compound 6a to demonstrate what happens due to our general opinion (Scheme 5) regardless of the type of the central cyclanone core in the structure.

The formation of dimer 20 from 6a under MALDI-TOF conditions (see experimental) in MS analysis. The related product ions of 6a observed: 289.127/[M-H]+; 291.172/[M + H]+; 290.126/[M] and the dimer 577.124/[M-3H]+; 580.119 [M]. In negative mode: 289.062/[M-H]−; 291.176/[M + H]−; 290.120/[M]
Mention must be made about the fact that it was not necessary to use a matrix. These curcuminoids are able to ionize themselves. They can behave as their own matrix in the presence of the laser shots.
There is a recent report on the unexpected formation of [M-H]+ ion during MALDI MS analysis of such cyclic C5-curcuminoids [37]. This ion can form under chemical experimental conditions [33,34,35,36] in the photochemical reaction too, together with the [M + H]− ion in a hydride transfer reaction. Therefore, it is not a surprise that the dimerization (2 + 2 cycloaddition) takes place. A very similar reaction is observable if we look at the proton transfer reaction. These two processes, hydride and/or proton transfer makes the cycloaddition possible. The molecular ion of dimeric compound 20 (or the dimeric fragment after loss of Cl or NO2 in the case of derivatives 18 or 6f) is there in the MALDI-TOF MS spectra of 6a. Similarly, all the product ions in Scheme 5 are there in the corresponding negative or positive ion mode records. The analytically useful diagnostic product ions are of importance for example when we analyze samples from complex biological matrices for identity or quantity of these C5-curcuminoids. That is why we propose to consider the dimeric ion or its fragment too for the mass spectrometric fingerprint analysis of these C5-curcuminoids in a MALDI-TOF technique for the future biological research.
Antiproliferative effects in vitro
The primary objective of the present work stems from our interest to study structure–activity relationships concerning these antiproliferative synthetic C5-curcuminoids. The biodata presented in Fig. 3 reveals that the majority of the newly described C5-curcuminiod derivatives exert pronounced antiproliferative activities as evaluated against the following three human adherent cancer cell lines: A2780 (ovarian), C33A (cervix) and MDA-MB-231 (breast). Data in Fig. 3 indicate the percentage of cell-growth-inhibition of the different cell lines in three different concentrations (0.3 μM, 3 μM and 30 μM) of the corresponding compound.

Evaluation of compounds 4–7, 15, 16, 18 and 19 against human A2780 (ovarian), C33A (cervix) and MDA-MB-231 (breast) cancer cell lines. The biodata reported here are percentage of cell-growth-inhibition in different concentrations. Cisplatin was used as a reference drug: IC50 = 1.30 (A2780); 3.69 (C33A); 19.13 (MDA-MB-231). Some selected IC50 calculated values (see also Table 2) = 6d: 2.46 (A2780), 1.38 (C33A), 1.71 (MDA-MB-231); 6p: 0.76 (A2780), 2.69 (C33A), 1.28 (MDA-MB-231); 6h: 0.68 (A2780), 0.69 (C33A), 0.92 (MDA-MB-231); 16 [18]: 0.22 (A2780), 0.43 (C33A), 0.37 (MDA-MB-231); 18c: 1.34 (A2780), 1.51 (C33A), 1.00 (MDA-MB-231). There was only negligible inhibition noted in all cases of compounds in 0.3 μM concentration except for compounds 18 and 19. Inhibition data of curcumin: A2780, 85% in 30 μM [39]; C33A, 22% in 30 μM [40]; MDA-MB-231, 50% in 30 μM [41]
Comparisons were made between the cytotoxic properties of compounds 4–7 (Fig. 2). Firstly, we can see a clear sequence among the structures in Fig. 2 regarding their activity. The heterocyclic derivatives 5a and 5b dominates the others in the case of all three cell lines. The second position is shared by the two homocyclic derivatives 6 and 7 favoring the hydroxyl-substituted compounds 6a and 6b in the case of the C33A (cervix) and A2780 (ovarian) cancer cells. The inhibition activity of the acyclic compound 4 in these two concentrations is negligible. Hence, the structural modifications in the ketone core of compounds 4–7 presented in Fig. 2 provided the following sequence in cell-growth-inhibition: N-methylpiperidones (5a, b) > 4-hydroxycyclohexanones (6a, b) > 4-methylcyclohexanones (7a, b) > acyclic ketone (4).
On the other hand, there is also a sequence in the structure–activity relation of the different aromatic substituents in series 6. There is a hydroxyl substituent in all the structures at position 4 of the newly synthesized 2,6-dibenzylidene-4-hydroxycyclohexanone 6a-m derivatives. The most active compounds are the nitro-substituted 6f (para), 6g (ortho) and 6h (meta) derivatives with nearly 100% inhibition in almost all cases even at 3 μM. The meta-nitro 6h counterpart shows the highest score, the other two isomers 6f and 6g are slightly weaker on C33A (cervix) cells. The second in this sequence is the pyridylidene substituent in derivatives 6o and 6p with better inhibition of the 4′-pyridyl derivative 6p on all three cell lines. The third position is occupied by the chloro substituent in compounds 6b (para), 6c (ortho,para–dichloro), 6d (meta) and 6e (ortho). The chloro-substituted subgroup is pharmacologically dominated by the meta-chloro 6d isomer with close to 100% inhibition on all cell lines. Interestingly, two of the most active cytotoxic derivative 6d and 6h has an electron-withdrawing meta-substituent in its structure. Their calculated IC50 values are as follows (see also Table 2): 6d: 2.46 (A2780), 1.38 (C33A), 1.71 (MDA-MB-231); 6h: 0.68 (A2780), 0.69 (C33A), 0.92 (MDA-MB-231).
Unexpectedly, the replacement of the previously mentioned substituents to methoxy group(s) in case of compounds 6j-m led to a lower inhibition percentage. These derivatives showed substancial inhibition only in the 30 μM concentration in almost all cases. Finally, the replacement of the para substituent on the aromatic rings by dimethylamino (6i), methyl (6n) or the exchange of the benzylidene groups to a longer aromatic obtained from cinnamaldehyde (6q), respectively, resulted in virtually inactive compounds. With the increase of the number of double bonds and the length of the spacer (from C5 up to C9) between the two aromatic rings in the structure of 6q the antiproliferative activity dropped to negligible.
These observations reveal that the electronic, hydrophobic and steric properties of the aryl substituents influence the magnitude of the inhibition ability of the compounds. The relative potencies of the most active compounds in series 6 with respect of the substituents on the aryl rings is meta-NO2 (6h) > 4′-pyridyl (6p) > meta-Cl (6d) > ortho-NO2 (6g) > para-NO2 (6f) > 3′-pyridyl (6o) > hydrogen (6a). Some selected IC50 calculated values (see also Table 2) - 6d: 2.46 (A2780), 1.38 (C33A), 1.71 (MDA-MB-231); Average: 1.85. 6p: 0.76 (A2780), 2.69 (C33A), 1.28 (MDA-MB-231); Average: 1.58. 6h: 0.68 (A2780), 0.69 (C33A), 0.92 (MDA-MB-231); Average: 0.76.
Only one of the members in series 6 was described earlier. Namely, compound 6f has been identified as a potent nonselective isopeptidase inhibitor intermediate in vivo against glioblastoma U87MG cells [42].
Derivatives of compound 6a were also prepared in order to change the structure of the secondary pharmacophore [28] at position 4 of the molecules with what the molecule binds to its biological place of action. Two structural modifications were accomplished on 6a: one of these acylation reactions provided the dimeric form 15 (Scheme 3) while the other one the chloroacetyl derivative 19 (Scheme 4). It was possible to increase the efficacy of 6a with the introduction of the chloroacetyl moiety into the structure and inhibition ability grew dramatically from the 30 μM concentration of 6a down to the 0.3 μM of 19. The difference in inhibition ability of compounds 5 and 6 (Fig. 2) could have been equalized due to this transformation from 6a to 19 comparing biodata of chloroacetyl derivatives 18a–d and 19. The ester type compound 19, according to biodata in Fig. 3, reached the relative inhibition potential of the corresponding heterocyclic amide analogues 18a–d on all the three cell cultures. The difference in their inhibition activity can be seen in the 0.3 μM concentration range, only (Fig. 3). The other attempt to modify the secondary binding part of 6a, however (Scheme 4), did not bring a positive change in antiproliferative action. The diester 15 proved to be less active compared to the analogously synthesized [18] diamide 16.
An evaluation of the biodata was undertaken to see if there was any selectivity of the compounds on the different cell cultures. The results obtained from the IC50 calculations of representative compounds (see the footnotes in Fig. 3) in this investigation shows there are differences in inhibition ability on the three cell lines. For example, compound 6h inhibits the cell-proliferation very actively in both A2780 and C33A, but it is less active in the case of MDA-MB-231 cells. Similarly, the most refractory cells against compound 6d were in the A2780 culture, and compound 6b showed the highest inhibition against the A2780 cell line. A similar tendency can be see among the derivatives with lower performances in the in vitro anticancer screening process. All in all, we can state there is a selectivity demonstrated by our compounds on the different tumor cell lines.
The reference compound of this study was cisplatin. The best performer compounds among the newly synthesized derivatives are 6d, 6p, 6h and 18c. Here we provide their calculated IC50 values (see also Table 2): cisplatin = 1.30 (A2780); 3.69 (C33A); 19.13 (MDA-MB-231); 6d = 2.46 (A2780), 1.38 (C33A), 1.71 (MDA-MB-231); 6p = 0.76 (A2780), 2.69 (C33A), 1.28 (MDA-MB-231); 6h = 0.68 (A2780), 0.69 (C33A), 0.92 (MDA-MB-231); 18c = 1.34 (A2780), 1.51 (C33A), 1.00 (MDA-MB-231). In the case of the MDA-MB-231 cancer cell line, all the new compounds exceeded the antiproliferative effect of cisplatin, just as in the case of the C33A tumor cells. The ovarian A2780 cell line showed a similar effect in case of compound 18c, lower activity with compound 6d while increased cell growth inhibition appeared in the case of 6p and 6h as compared to the reference compound.
Inhibition percentage data of curcumin are known from the literature: A2780, 85% in 30 μM [39]; C33A, 22% in 30 μM [40]; MDA-MB-231, 50% in 30 μM [41]. We can state, that the majority of the new compounds in series 6, 18 and derivative 15 and 19 are exceeding these values of curcumin. The compounds with lower antiproliferative activity are 6i, 6j and 6q in A2780 culture, 6i and 6q in C33A while 6i and 6q in MDA-MB-231.
DNA binding
CD is a reliable tool for the detection of DNA binding of ligands. The appearance of the induced circular dichroism signal (ICD) is definitive proof of the interaction. In addition, shifts in the DNA bands are also indicative of the binding [48, 49].
There is little known about the DNA binding of cyclic C5-curcuminoids. About curcumin, however, we know more in this respect. It is known from in vitro experiments, for example, that curcumin appears in the nucleus of cultured glioma cells after incubation. It was revealed that nuclear homing is not a result of curcumin’s DNA binding [27]. The temporal relationship of curcumin’s apoptotic induction effect and its nuclear homing is under investigation to acquire details about the mechanism of action. This fact among others prompted us to initiate measurements to see possible interactions between C5-curcuminoids and DNA. For this reason, we conducted circular dichroism spectroscopic investigations on the newly synthesized cyclic C5-curcuminoid derivatives in series 6 and 18 using natural DNA.
Curcumin, as well as 28 C5-curcuminoid derivatives were tested on chicken erythrocyte DNS. In the case of curcumin, a large ICD band appeared in the ligand’s absorption region, showing strong interaction between the molecule and the polynucleotide (Fig. 4). It also caused some minor shift in the DNA band, meaning the double helical structure is slightly distorted (Fig. 4). Out of the tested C5-curcuminoid derivatives (6, 18), those containing either aliphatic or aromatic nitrogen showed signs of interaction—a weak ICD sign appeared in the recorded spectra for example in the case of 6i (Fig. 4). DNA bands did not change significantly in the same time, indicating that the polynucleotide remains in its native B-form. The binding is most probably the result of the ionic bondage of the nitrogen atoms in the ligand structure with the phosphate groups of DNA. In the case of other derivatives neither an ICD signal, nor shift in the DNA bands were detected, marking that no interaction occurs between the molecules (e.g. compounds 6h and 18c in Fig. 4).

CD and UV titration of 0.033 mg/ml chicken erythrocyte DNA (black curve) with 5 mM curcumin or curcuminoid stock solution (final concentrations are: 16.66 µg/mL, 25 µg/mL and 33.33 µg/mL, respectively)
Although it would be consistent with an ability of these curcuminoids to adopt a close-to-planar molecular shape, none of them showed stronger interaction with the DNA used in this study. It would be conceivable that the very active inhibitor 6h or 18c show diverse interaction comparing to the practically ineffective 6i. However, compounds in series 6 and 18 showed similar properties under CD conditions, that makes possible to generalize: based on these data we conclude that these derivatives do not bind to DNA in vitro. Hence, their antiproliferative effect is not due to their interaction with DNA.
Physicochemical calculations on derivatives of series 6
For an evaluation of some physicochemical properties compared to curcumin the synthesized compounds were computed. ChemAxon’s Marvin Suite was used for all of the calculations [43].
The most promising compound from physicochemical point of view (Table 2) is the 4′-pyridylidene derivative 6p. This compound is one of those, which revealed the highest cytotoxic activity (6d, 6p, 6h and 18c) against the cancer cell lines, with a relatively low partition coefficient (ClogP), with the possibility for ionization and with the highest water-solubility. Its ionization is due to the presence of the two pyridine nitrogen atoms which can be protonated under physiological conditions. The protonation can increase the solubility in water and decrease lypophilicity resulting in a possible better penetration through membranes into biological tissues, such as tumors. These data about this drug-candidate 6p are suggesting its possible higher bioavailability compared to curcumin consistent with Lipinski rules [44]. Similar physicochemical properties could be determined on the 3′-pyridylidene counterpart 6o too.
In general, the molecular weight of the newly synthesized derivatives are in the range of that of curcumin except the highly chlorinated 6c, 18b, the methoxy or dimethylamino substituted 6k, 18c, 18d and the dimer 15. The calculated partition coefficient (ClogP) values are usually under 5 (in the majority of cases between 3 and 4) or nearly 5 (6q, 18b, 19), except chlorinated compounds 6b-e and the para-tolyl substituted 6n. The moderately active dimeric derivative 15 exhibited an extremely high ClogP value of 9.505.
Those compounds with a slightly basic nitrogen (6i, 6o, 6p and 18d) or with a phenolic OH group (6m) on the arylidene substituents are slightly less lypophilic than curcumin. The reason for this diminished lypophilicity can be protonation or deprotonation. The calculated water-solubility value, however, is increased only in the case of pyridylidene derivatives 6o and 6p. All of the compounds in Table 2 are able to form 3–7 H-bonds giving the chance to develop more or less weak interactions with macromolecules on the place of biological action. There is no visible physicochemical reason on the practical ineffectiveness of compounds 6i and 6q. Further investigations about the relationship of biological properties with physicochemical behaviors will be undertaken in the near future in our laboratories.
Conclusions
A newly designed branch of bis(arylidene)-4-cyclanones is described here in this article. The structure of this new group is based on 4-hydroxycyclohexanone, which was converted to the title-compounds, 2,6-bis(arylidene)-4-hydroxycyclohexanone (6) species under the conditions of the Claisen-Schmidt condensation reaction. Series 6 and its derivatives 15, 18, 19 were evaluated against three malignant experimental cancer cell lines in vitro: A2780 (ovarian), C33A (cervix) and MDA-MB-231 (breast). Antiproliferative biodata were obtained beside cisplatin as a reference drug in the form of tumor-growth-inhibition percentage in three different concentrations. Structure–activity analysis revealed, that the best performer in this study is (E,E)-2,6-bis(3′-nitrobenzylidene)-4-hydroxycyclohexanone (6h) with IC50 values of 0.68, 0.69 and 0.92 μM, respectively, compared to cisplatin with 1.30, 3.69 and 19.13 μM. Calculations were performed on computed compounds in order to obtain a relationship/correlation between biodata and physicochemical parameters. The best performer in this respect was 6p and 6o, in which case it was possible to predict improved bioavailability compared to curcumin. Acylation of the functional group at position 4 with chloroacetyl chloride was successful from pharmacological point of view. It led us to very effective acyl derivatives. It was possible for instance to increase the efficacy of 6a with the introduction of the chloroacetyl moiety into the structure of compound 19: inhibition ability grew dramatically from the 30 μM concentration down to the 0.3 μM. We have selected some of these derivatives for CD spectroscopic investigation in order to see whether they show any interaction to natural DNA. Based on our CD spectroscopic data, we conclude that these derivatives do not bind to DNA in vitro. Their antiproliferative effect is not due to their interaction with DNA.
References
World Health Organization: World Cancer Report 2018, http://www.who.int/mediacentre/factsheets/fs297/en
F.E. Koehn, G.T. Carter, Nat. Rev. Drug Discov. 4, 206 (2005)
A.S. Oliveira, E. Sousa, M.H. Vasconcelos, M. Pinto, Curr. Med. Chem. 22, 4196 (2015)
B.B. Aggarwal, A. Kumar, A.C. Bharti, Anticancer Res. 23, 363 (2003)
T. Masuda, A. Jitoe, J. Isobe, N. Nakatani, S. Yonemori, Phytochemistry 32, 1557 (1993)
J.L. Jiang, X.L. Jin, H. Zhang, X. Su, B. Qiao, Y.J. Yuan, J. Pharm. Biomed. Anal. 70, 664 (2012)
A. Kohyama, H. Yamakoshi, S. Hongo, N. Kanoh, H. Shibata, Y. Iwabuchi, Molecules 20, 15374 (2015)
K. Selvendiran, S. Ahmed, A. Dayton, M.L. Kuppusamy, M. Tazi, A. Bratasz, L. Tong, B.K. Rivera, T. Kálai, K. Hideg, P. Kuppusamy, Free Radic. Biol. Med. 48, 1228 (2010)
A. Dayton, K. Selvendiran, M.L. Kuppusamy, B.K. Rivera, S. Meduru, T. Kálai, K. Hideg, P. Kuppusamy, Cancer Biol. Ther. 10, 1027 (2010)
T. Kálai, M.L. Kuppusamy, M. Balog, K. Selvendiran, B.K. Rivera, P. Kuppusamy, K. Hideg, J. Med. Chem. 54, 5414 (2011)
P. Lagisetti, P. Vilekar, K. Sahoo, S. Anant, V. Awasthi, Bioorg. Med. Chem. 18, 6109 (2010)
A. Jha, C. Mukherjee, A.K. Prasad, V.S. Parmar, E. De Clerck, J. Balzarini, J.P. Stables, E.K. Manavathu, A. Shrivastav, R.K. Sharma, K.H. Nienaber, G.A. Zello, J.R. Dimmock, Bioorg. Med. Chem. 15, 5854 (2007)
H.N. Pati, U. Das, S. Das, B. Bandy, E. De Clerck, J. Balzarini, M. Kawase, H. Sakagami, J.W. Quail, J.P. Stables, J.R. Dimmock, Eur. J. Med. Chem. 44, 54 (2009)
H.N. Pati, U. Das, J.W. Quail, M. Kawase, H. Sakagami, J.R. Dimmock, Eur. J. Med. Chem. 43, 1 (2008)
N. Li, W. Xin, B. Yao, C. Wang, W. Cong, F. Zhao, H. Li, Y. Hou, Q. Meng, G. Hou, Eur. J. Med. Chem. 147, 21 (2018)
U. Das, H. Sakagami, Q. Chu, Q. Wang, M. Kawase, P. Selvakumar, R.K. Sharma, J.R. Dimmock, Bioorg. Med. Chem. Lett. 20, 912 (2010)
Q. Chen, Y. Hou, G. Hou, J. Sun, N. Li, W. Cong, F. Zhao, H. Li, C. Wang, Res. Chem. Intermed. 42, 8119 (2016)
S. Das, U. Das, A. Varela-Ramirez, C. Lema, R.J. Aguilera, J. Balzarini, E. De Clerck, S.G. Dimmock, D.K.J. Gorecki, J.R. Dimmock, ChemMedChem 6, 1892 (2011)
K. Selvendiran, L. Tong, S. Vishwanath, A. Bratasz, N.J. Trigg, V.K. Kutala, K. Hideg, P. Kuppusamy, J. Biol. Chem. 282, 28609 (2007)
U. Das, H.N. Pati, Z. Baráth, Á. Csonka, J. Molnár, J.R. Dimmock, Bioorg. Med. Chem. Lett. 26, 1319 (2016)
S. Das, U. Das, P. Selvakumar, R.K. Sharma, J. Balzarini, E. De Clercq, J. Molnár, J. Serly, Z. Baráth, G. Schatte, B. Bandy, D.K. Gorecki, J.R. Dimmock, ChemMedChem 4, 1831 (2009)
X. Yuan, H. Li, H. Bai, Z. Su, Q. Xiang, C. Wang, B. Zhao, Y. Zhang, Q. Zhang, Y. Chu, Y. Huang, Eur. J. Med. Chem. 77, 223 (2014)
U. Das, R.K. Sharma, J.R. Dimmock, 1,5-Diaryl-3-oxo-1,4-pentadienes: curr. Med. Chem. 16, 2001 (2009)
E. Burgos-Morón, J.M. Calderón-Montano, J. Salvador, A. Robles, M. López-Lázaro, Int. J. Cancer 126, 1771 (2010)
B.T. Kurien, S.P. Dillon, Y. Dorri, A. D’Souza, R.H. Scofield, Int. J. Cancer 128, 239 (2011)
F. Zsila, Z. Bikadi, M. Simonyi, Org. Biomol. Chem. 2, 2902 (2004)
M. Ghosh, R.O. Ryan, J. Nutritional Biochem. 25, 1117 (2014)
I. Huber, I. Zupkó, I.J. Kovács, R. Minorics, G. Gulyás-Fekete, G. Maász, P. Perjési, Monatsch. Chem. 146, 973 (2015)
J.R. Dimmock, M.P. Padmanilayam, G.A. Zello, K.H. Nienaber, T.M. Allen, C.L. Santos, E. De Clercq, J. Balzarini, E.K. Manavathu, J.P. Stables, Eur. J. Med. Chem. 38, 169 (2003)
P.N. Craig, J. Med. Chem. 14, 680 (1971)
C. Hansch, A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology (Wiley, New York, 1979), p. 75
P. Sohár, A. Csámpai, P. Perjési, Arkivoc 5, 114 (2003)
E. Diaz, H. Barrios, D. Corona, A. Guzmán, R. Diaz, A. Fuentes, Spectrochim. Acta, Part A 58, 2079 (2002)
G. Kaupp, M. Plagmann, J. Photochem. Photobiol. A: Chem. 80, 399 (1994)
G. Kishore, K. Kishore, Macromolecules 26, 2995 (1993)
G.C. Forward, D.A. Whiting, J. Chem. Soc. (C) 1868 (1969)
H. Awad, M.J. Stoudemayer, L. Usher, I.J. Amster, A. Cohen, U. Das, R.M. Whittal, J. Dimmock, A. El-Aneed, J. Mass Spectrom. 49, 1139 (2014)
J.B. Aldersley, G.N. Burkhardt, A.E. Gillem, N.C. Hindley, J. Chem. Soc. (C) 10 (1940)
L. Zheng, Q. Tong, C. Wu, Chin. J. Integr. Med. 12, 126 (2006)
S. Shukla, S. Mahata, G. Shishodia, A. Pandey, A. Tyagi, PLoS ONE 8(7), e67849 (2013). https://doi.org/10.1371/journal.pone.0067849
C.P. Prasad, G. Rath, S. Mathur, D. Bhatnagar, R. Ralhan, Chem. Biol. Interact. 181, 263 (2009)
U. Cersosimo, A. Sgorbissa, C. Foti, S. Drioli, R. Angelica, A. Tomasella, R. Picco, M.S. Semrau, P. Storici, F. Benedetti, F. Berti, C. Brancolini, J. Med. Chem. 58, 1691 (2015)
Calculator Plugins were used for structure property prediction and calculation, Marvin suite 15.2.23, ChemAxon. http://www.chemaxon.com
C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Adv. Drug Deliv. Rev. 46, 3 (2001)
A. Arnold, M. Markert, R. Mahrwald, Synthesis 38, 1099 (2006)
J. Deli, T. Lóránd, D. Szabó, A. Földesi, Pharmazie 39, 539 (1984)
G.H. Mahdavinia, M. Mirzazadeh, J. Chem. 9, 49 (2012)
S. Allenmark, Chirality 15(5), 409 (2003)
E. Kiss, A. Mirzahosseini, A. Hubert, A. Ambrus, L. Őrfi, P. Horváth, J. Pharm. Biomed. Anal. 150, 355 (2018)
T. Mosmann, J. Immunol. Methods 65, 55 (1983)
Acknowledgements
Open access funding provided by University of Pécs (PTE). This work was supported by the University of Pécs, Faculty of Medicine Research Fund PTE ÁOK-KA-34039-12/10-11.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Huber, I., Zupkó, I., Gyovai, A. et al. A novel cluster of C5-curcuminoids: design, synthesis, in vitro antiproliferative activity and DNA binding of bis(arylidene)-4-cyclanone derivatives based on 4-hydroxycyclohexanone scaffold. Res Chem Intermed 45, 4711–4735 (2019). https://doi.org/10.1007/s11164-019-03859-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-019-03859-4