
Abstract
In recent years, the number of tools and techniques that enable genetic material to be added, removed or altered at specific locations in the genome has increased significantly. The objective is to know the structure of genomes, the function of genes and improve gene therapy.
In this work we intend to explain the functioning of the CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9) and the advantages that this technique may have compared to previously developed techniques, such as RNA interference (RNAi), Zinc Finger Nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) in gene and genome editing.
We will start with the story of the discovery, then its biological function in the adaptive immune system of bacteria against bacteriophage attack, and ending with a description of the mechanism of action and its use in gene editing. We will also discuss other Cas enzymes with great potential for use in genome editing as an alternative to Cas9.
CRISPR/Cas9 is a simple, inexpensive, and effective technique for gene editing with multiple applications from the development of functional genomics and epigenetics. This technique will, in the near future, have great applications in the development of cell models for use in medical and pharmaceutical processes, in targeted therapy, and improvement of agricultural and environmental species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Genes editing techniques
Before the appearance of the CRISPR technique, tools such as interference RNAs, ZFNs (zinc finger nucleases), and TALENs (Transcription activator-like effector nucleases) had already been developed and utilized in genes editing.
RNA interference (RNAi)
The RNA interference tool (RNAi) was the first to be developed and has become widely used for experimental use. However, it was only in 1998, thanks to the study carried out by Andrew Fire and Craig Mello [1], these researchers revealed that long double-stranded RNA molecules (dsRNA) containing part of the target gene were able to specifically inhibit the expression of this gene by decreasing the target mRNA. RNAi is a small molecule, its siRNA synthesis is cheap and the transfection methods (electroporation, lipofection) are quite efficient; these characteristics allow an increased number of cell types and organisms where this technique can be used. Furthermore, different researchers have already investigated the therapeutic potential of these molecules, the most prominent example being the use of the RNAi tool for the treatment of familial amyloidosis caused by transthyretin [2]. Despite some of the advantages of the RNAi technique mentioned above, it also has disadvantages that have been noticed over the years, one of which was discovered in 2007 by Krueger and colleagues [3] who observed that gene silencing was often not complete, thus producing a knockdown (not knockout) effect.
Zinc finger nucleases
Nucleases are another tool widely used in genomics that allows the manipulation and alteration of the nucleotide sequence in a specific way. Nucleases are capable of causing a break in the double strand of DNA, instigating repair pathways by NHEJ (non-homologous end joining) or (b) HDR (homology-directed repair) and enabling the insertion/deletion of bases and homologous recombination with the donor DNA. ZFNs were the first nucleases capable of causing an alteration in a supervised manner in a predetermined location in the genome and thanks to this, the insertion of the Il2RG gene in X-linked immunodeficiency models (SCID-X1) in vitro has already been successfully performed [4] and in vivo [5]. However, ZFN technology requires complex engineering in the design and assembly of zinc finger domains, in addition to specificity problems. Not all produced ZFNs have the ability to effectively cleave DNA, and possibly guanine-containing target sites in the 5’ region have a higher success rate [6].
Transcription activator-like effector nucleases (TALENs)
The genomic tool TALEN was described in 2009 by several researchers [7, 8] and presents several similarities with ZFNs, as both use the same FokI domain to cleave the target site. This tool has the ability to bind to DNA due to the presence of domains such as transcription activator-like effectors (TALEs) in its structural composition. These domains originate from a pathogen that attacks plants, the bacterium Xanthomonas spp [9]. However, numerous studies have shown that the editing efficiency of TALENs is relatively lower than that of CRISPR, with TALENs being approximately 1 to 60% and CRISPR (2.3 to 79%) [10,11,12,13,14].
There are numerous advantages in using CRISPR over the other genomic tools mentioned above. CRISPR is simple and easy to design the gRNA (only about 20 nucleotides that will compose the guide RNA and that will be complementary to the region to be targeted) [10]. In addition, CRISPR has a greater mutation efficiency and allows for the manipulation of more than one gene at the same time, thus making it possible to originate multiple mutations [15,16,17], without the off-target effects produced by TALENs and ZFNs [18, 19].
Contextualization
Currently, the technique of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) has has been widely discussed, both in the scientific community and in the media in general. This is due to the fact that this technique is triggering a revolution in molecular genetics, because it allows, among other functions, genetic editing, that is, the ability to alter the nucleotide sequence. This alteration can be of different types: (I) deletion, insertion, or substitution of one or more nucleotides, (II) integration of genetic elements or (III) deletion of genetic elements. In addition, CRISPR can also be used for purposes other than gene editing, for example: (I) DNA tagging, (II) regulation of gene expression, (III) RNA cleavage, (IV) gene mapping, or (V) RNA tracking.
According to Timlin et al., 2018, the use of the CRISPR / Cas9 (CRISPR associated protein 9) technique makes it unnecessary to resort to protein engineering to develop a site-specific nuclease against a specific target DNA sequence. All it takes is the synthesis of a new piece of RNA, thus reducing the time required for the design and implementation of gene editing.
History
In 1987, the researcher Yoshizumi Ishino and his collaborators identified a locus (region) that aroused particular interest in the genome of Escherichia coli bacteria. This locus was organized in an unusual configuration with repeated sequences and intercalated spacer sequences and of unknown function [21]. In addition to researcher Yoshizumi Ishino, in 1993 and 2000, other researchers [22, 23, 24] also carried out the identification of these sequences in the genomes of different bacteria and Archaea. However, it was only in 2002 that the acronym CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) was formulated to refer to such sequences [25]. Subsequently, a set of genes physically very close to the CRISPR locus were also identified and named Cas genes (CRISPR associated genes) [25], genes that would be recognized as central elements in the functioning of the locus as a whole.
In 2005, three independent groups of researchers demonstrated that spacer sequences have an extrachromosomal origin, that is, they are sequences that are derived from plasmids or viruses. Later, it was also described that viruses are unable to successfully infect bacteria that have spacers whose sequences correspond to stretches of their genomes [26, 27, 28].
As a result of these findings, it was hypothesized that CRISPR-Cas would be an adaptive immune system of prokaryotes and in which spacers would serve as “memory of previous invasions” [27]. Thus, RNA molecules produced from these spacers would be complementary to the (re)invasive pathogen, making it possible to fight it in a sequence-specific manner. A series of subsequent studies revealed that this hypothesis was indeed correct. According to [29], this “memory” could be used by Cas proteins, helping as guided endonucleases, with the objective of scanning the invading DNA and deactivating it by introducing double-stranded breaks (DSBs).
The biological function of CRISPR-Cas
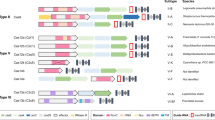
The CRISPR-Cas system is an adaptive defense system that allows prokaryotes to protect themselves against a possible (re)invasion of unwanted mobile genetic elements, such as bacteriophages, transposons, and plasmids [30]. In this system, immunity is mediated by Cas nucleases and small guide RNAs crRNAs (CRISPR-derived RNA), which distinguish the cleavage site within the invading genome. This system is made up of two main components: the Cas proteins, which act as catalysts, and the CRISPR locus, which functions as genetic memory. The defense mechanism consists of three steps: (I) adaptation, (II) crRNA biogenesis, and (III) action against the invader, as represented in Fig. 1.

(a) The three steps of defense mechanism: (I) adaptation, (II) crRNA biogenesis, and (III) Targeting. (b) The different types of CRISPR-Cas systems (Type I, Type II, and Type III) [36]
The first stage consists of a process of adaptation and is characterized by the acquisition of a new spacer at the CRISPR locus [31]. This process occurs, for example, when the bacteria is first infected by the virus. At this point, some enzymes encoded by the Cas genes (Cas1 and Cas2) cleave the pathogen’s DNA into small segments (24–48 base pairs) and integrates them into the CRISPR locus as new SPACERS (i.e., between the repeated sequences). From that moment on, the bacterium is immunized against future invasions by this same agent.
The second step, crRNA biogenesis, is the uninterrupted transcription of the CRISPR locus. This process is mediated by the lead sequence (L), a region with a high adenine-thymine content that serves as a promoter site. Transcription from this locus generates a “CRISPR RNA precursor” (or pre-crRNA), containing multiple repeat sequences and multiple spacers in a single long RNA. Subsequently, this pre-crRNA will be processed, giving rise to several smaller RNAs, the crRNAs, each corresponding to a different spacer [31].
In the third step, Targeting, mature crRNAs together with Cas proteins, form complexes that recognize the exogenous genetic sequence (plasmid, transposon, or virus) and destroy it [31] by introducing DSB [29]. This process recalls some similarities with the RNA Interference (RNAi) mechanism observed in eukaryotes [32, 33].
The system elements vary between different species, both in terms of occurrence, gene composition, sequences, number, and size [34]. For example, in the case of the composition of spacers, they can even vary between the same species, as they depend on previous contacts they have with different pathogens. Researches have revealed the importance of this mechanism for applications, namely: bacterial resistance to phages; control of gene dissemination; strain genotyping (based on spacer hypervariability), and the study of microbial population dynamics [35]. In addition, there are already several studies using this machinery as an effective form of genetic engineering, not only in microorganisms but also in the case of plants of economic interest [36].
Three different types of CRISPR-Cas system (Type I, Type II, and Type III) have been identified [36] and were grouped according to the conservation of cas genes and operon organization [37], see Fig. 1(b). In the case of CRISPR Type I and Type III systems, they use sets of Cas proteins. The Type I system uses a multiprotein CRISPR RNA (crRNA) complex known as Cascade, this complex recognizes the target DNA, which is then cleaved by Cas3. For the Type III system, Cas10 is assembled into a Cascade-like complex that recognizes and cleaves the target [36, 38]. Regarding the CRISPR-Cas Type II system, it can be defined by the presence of the endonuclease Cas9, guided by the crRNA. In this system, the cas9 gene is the only one needed to fight the invading DNA. During an infection, the complex formed by Cas9 destroys the viral genome, also relying on the protospacer adjacent motif (PAM) domain. The Cas9 enzyme contains two domains with nuclease activities (RuvC and HNH), which participate in immunity [31, 39].
CRISPR/CAS 9
According to [35], the CRISPR-Cas system occurs in nature and can be defined as a prokaryotic immunity system based on the capture and insertion of small DNA fragments coming from the invasion by viruses or plasmids. These fragments are later incorporated into the bacterium’s genome and against which it then acquires resistance. Taking advantage of this natural biological process, the researchers managed to develop the technique called CRISPR-Cas9, this technique uses a guide RNA (thus referring to CRISPR) and only one of the Cas proteins (the endonuclease Cas9). In order to simplify the names, many investigators refer to the technique simply as CRISPR. However, it is important to note that the endonuclease used is not always Cas9, other endonucleases such as Cpf1 have been used recently (CRISPR-Cpf1) [40].
The endonuclease most frequently used in eukaryotic cell DNA editing is the enzyme (SpCas9), and it comes from the bacterial species Streptococcus pyogenes. The three-dimensional analysis of this enzyme revealed that it has a bilobed structure: a recognition lobe (REC) and a lobe with nuclease activity (NUC). The NUC lobe has the catalytic domains RuvC and HNH, in addition to the PAM interaction domain (PI) [41].
In order for the Cas9 enzyme to perform its function, it needs to be activated and targeted to its target. In the process of natural immunity in bacteria, these steps are aided by two cooperatively acting RNA molecules: crRNA and tracrRNA (trans-activating RNA) as in Fig. 2(a). However, in order to make this process as simple as possible for laboratory application, the researchers developed the single guide RNA (sgRNA or gRNA): a chimeric molecule resulting from the “fusion” of crRNA and tracrRNA, synthesized to accumulate the two functions, which are highly dependent on their structures [41]. Basically, the CRISPR technique involves three molecules: a nuclease (usually the wild-type Cas9 from S. pyogenes), a guide RNA (known as single guide RNA), and the target (often DNA), thus facilitating the experimental procedure.
The sgRNA is formed by a hairpin fold consisting of the target recognition sequence also known as the guide sequence (∼20 nt at the 5’ end, specific for each target) plus a universal sequence (∼80 nt, the scaffold, end 3´) and conserves the base-pairing interactions in the double-strand [41] See Fig. 2(b). At first, sgRNA was tested in prokaryotes [41, 42], but more recently it has been used extensively in editing mammalian cell genomes [42].
The region in the DNA that will be cleaved by the Cas9 nuclease has two elements: (I) the target itself and (II) a PAM sequence [43, 44]. The Cas9/sgRNA complex will interact with the target only if there is an adjacent PAM motif on the other strand of DNA. PAMs are short sequences, usually 2–5 nt (e.g., 5’NGG3’ and 5’NNGRRT3’), essential for anchoring the nuclease to the cleavage site.

(a) The process of natural immunity in bacteria, these steps are aided by two cooperatively acting RNA molecules: crRNA (CRISPR-derived RNA) and tracrRNA (trans-activating RNA). (b) (sgRNA or gRNA): a chimeric molecule resulting from the “fusion” of crRNA and tracrRNA, synthesized to accumulate the two functions [20]
Cas9/sgRNA mechanism of action
Double cut cleavage
Crystallographic investigations of the SpCas9 enzyme have highlighted some of the main steps in the cleavage process.
Initially, the interaction of Cas9 with sgRNA results in an alteration in the protein’s structure. This change facilitates the later interaction between the PAM Interacting (PI) domain and the PAM sequence. In this configuration, PI acts simultaneously on the two strands of the target DNA, separating them.
This process occurs as follows: conserved arginine residues in PI bind to the dinucleotide “GG” of the PAM motif (5’NGG3’), spatially limiting the movement of the non-target chain. Simultaneously, another region of PI interacts with a phosphodiester group in the target chain, “pulling it away” from the non-target chain. This action results in the local separation of the target DNA strands (strand opening) immediately upstream of the PAM sequence [45]. This local opening allows the start of the interaction of the guide sequence (from the sgRNA) with the target itself, which occurs just upstream of PAM [45, 46]. The initial interaction step involves a fundamental domain of the guide sequence called seed, ~ 8 nt in length [47]. From the seed, the pairing between sgRNA and the target is initiated, however, bad pairings between seed and a possible target are not allowed, thus preventing the stabilization of Cas9/sgRNA with this molecule. Otherwise, with perfect pairing between seed and target, the opening process (separation of the two strands of DNA) proceeds, resulting in a stable interaction between Cas9/sgRNA and the target. Ultimately, the negative charge of the sgRNA/DNA-target heteroduplex is accommodated in the groove formed at the interface between the REC and NUC lobes of Cas9, which have a positive charge [45]. From that moment on, Cas9 is able to cleave the target in the two strands of DNA, through its catalytic domains HNH (which cleaves the target strand, complementary to the guide) and RuvC (which cleaves the other strand). This cleavage occurs within the target, about 3–4 bp upstream of PAM, generating blunt ends [41].
Single cut cleavage
In addition to the double-cut cleavage, it is still important to mention that a variant of the wild type (i.e., unmodified) Cas9 enzyme, named Cas9 nickase (Cas9n), has recently been developed [10]. This variant allows cutting only one of the two strands of the target DNA and this occurs due to the inactivation of one of its catalytic domains (RuvC or HNH) [41, 38, 49].
spCas9, saCas9, and Cpf1 nucleases
To overcome the limitations of spCas9, such as its large size and G-rich PAM sequence, other CRISPR enzymes have been alternatively proposed, the most mentioned being Cpf1 and saCas9.
Despite its versatility, the size of the S. pyogenes Cas9 (SpCas9) limits its usefulness for basic research and therapeutic applications using the highly versatile adeno-associated virus (AAV) delivery vehicle. Ran et al., in 2015 [50] characterized six smaller Cas9 orthologs and they showed that Staphylococcus aureus Cas9 (SaCas9) can edit the genome with similar efficiencies to SpCas9, although being more than 1 kilobase shorter. In 2015, Ran and his collaborators, can packaged SaCas9 and its unique guide RNA expression cassette into a single AAV vector and targeted the cholesterol-regulating gene Pcsk9 in mouse liver. One week after injection, they observed > 40% gene modification, accompanied by significant reductions in serum Pcsk9 and total cholesterol levels [50].
A new CRISPR nuclease, Cpf1, was discovered in late 2015 by Feng Zhang’s group at MIT [51].
Cpf1 allows for new targeting possibilities as it recognizes T-rich PAM sites. While Cas9 may be the preferred nuclease for targeting G-rich areas, Cpf1 can be used to target T-rich areas. Cpf1 requires a shorter guide RNA to operate. While Cas9 requires the presence of a tracrRNA to process crRNA, Cpf1 can process pre-crRNA by itself [25]. This is of particular interest for biotechnology, as compared to ~ 100 nt tracrRNA/crRNA hybrids used with Cas9, Cpf1 can be targeted with only a ~ 42 nt crRNA. This reduces the size of the designed sgRNA by more than half, while simplifying the methods and costs associated with synthesis and (if desired) chemical modification [52].
Another notable difference is that Cas9 generates blunt ends after cleavage, while Cpf1 leaves 5’ sticky bumps that can be used for directional cloning.
Cpf1 cleaves DNA 18–23 bp downstream of the PAM site, resulting in no disruption to the recognition sequence after NHEJ repair of double-stranded DNA break. As a result, Cpf1 allows for multiple rounds of DNA cleavage and a greater opportunity for the desired genomic editing to occur [53].
DNA repair: NHEJ and HDR
The cleavages performed by Cas9, generating sharp ends (cut in both strands) or nicks (cut in only one of the strands), must be repaired by the cell. In this scenario, two mechanisms can come into play: (a) NHEJ (non-homologous end joining) or (b) HDR (homology-directed repair) [20] as represented in Fig. 3.

The two mechanisms of DNA repair: (a) NHEJ (non-homologous end joining) and (b) HDR (homology-directed repair) [20]
NHEJ
In general, and simplifying the process, this mechanism has the function of bringing together the two ends of DNA, through events that do not depend on homology between the two ends. However, this pathway is intrinsically error-prone, generating Indel-type mutations (insertion or deletion of one or a few nucleotides) at or near the tip junction site [42]. These mutations, in turn, will compromise the functionality of the target gene’s end product. That is, the joint action of Cas9-NHEJ often results in gene knockout. Gene knockout is of great interest for genetics, cell biology, biomedicine, and several other areas, with several applications.
HDR
When the ends generated by the cleavage have homology to each other (or to some third molecule), there is another repair pathway that can be recruited – HDR, based on homologous recombination. Sometimes the cleavage of target DNA by the Cas9 enzyme does not generate ends with some degree of homology. However, even in these situations the HDR repair process can be triggered. For this, it is necessary to introduce a donor DNA molecule, which must have homology with both ends resulting from the cut.
The use of HDR repair is essential when the goal is to generate “knock-in” cells/organisms or to perform an allelic replacement. Knock-in generally refers to the introduction of a new DNA sequence into the genome, such as a transgene [54]. Allelic substitution, on the other hand, refers to the exchange of a nucleotide sequence for another that is very similar, but distinct (i.e., of one allele for another) [55]. In both cases, the CRISPR experimental procedure is altered, that is, three exogenous elements are introduced into the cell: Cas9 nuclease, sgRNA, and donor DNA (the trigger of HDR).
Cas Nucleases future trends
Wang JY et al. [56] found that in some CRISPR systems, a fusion of reverse transcriptase (RT) with Cas1 integrase and Cas6 maturase creates a single protein that allows for sequence-matched integration and crRNA production. To elucidate how RT-integrase organizes distinct enzymatic activities, they present the cryo-EM structure of a Cas6-RT-Cas1-Cas2 CRISPR integrase complex. The structure reveals a heterohexamer in which the RT directly contacts the integrase and maturase domains, suggesting functional coordination between all three active sites. These findings highlight an expanded ability of some CRISPR systems to acquire diverse sequences that drive CRISPR-mediated interference [56].
Liu et al., [57] revealed the underlying mechanisms of a distinct third RNA-guided genome editing platform called CRISPR – CasX, which uses unique structures for programmable double-stranded DNA ligation and cleavage. With biochemical and in vivo data, they demonstrated that CasX was active for Escherichia coli and human genome modification. They demonstrated how CasX activity arose through convergent evolution to establish a family of enzymes that is functionally separate from Cas9 and Cas12a [57].
Pausch et al. [58], described a minimally functional CRISPR-Cas system comprising a single ~ 70 kilodalton protein, CasΦ, and a CRISPR matrix, encoded exclusively in the genomes of huge bacteriophages. CasΦ uses a single active site for CRISPR RNA processing and crRNA-guided DNA cutting to target foreign nucleic acids. This hypercompact system, according to these authors, is active in vitro and in human and plant cells with expanded target recognition capabilities compared to other CRISPR-Cas proteins. Useful for genome editing and DNA detection, but with a molecular weight half that of Cas9 and Cas12a genome editing enzymes, CasΦ offers advantages for cell delivery that expand the genome editing toolbox [58].
The CRISPR-Cas Type VI system contains the programmable single effector guided RNA with RNases Cas13 [59]. CRISPR-Cas13 can be used as a flexible platform to study RNA in mammalian cells and for therapeutic development [60].
The diagnostic tool, SHERLOCK (Specific Enzymatic Reporter UnLOCKing) developed by Biosciences, uses synthetic RNA strands to create a signal after cutting and Cas13 will cut this RNA after cutting its original target, releasing the signaling molecule.
In 2020, the Food and Drug Administration granted authorization for a CRISPR-based test that can diagnose COVID-19 in about an hour. The test uses the Cas13a enzyme to identify an RNA sequence unique to SARS-CoV-2.
Conclusions
As early as 1985 Oliver Smithies and Mario Capecchi carried out work involving the integration of a donor DNA via homologous recombination [61], that is, the beginning of gene editing; however, only after three decades did the terms gene editing or editing of genes gain attention from the general population. Much of this attention was mainly due to the more widespread use of nucleases that allow target-site cleavage (e.g., ZFNs [62], TALENs [63], and CRISPR/Cas9), allowing the generation of DBS and activating the repair systems, thus dramatically increasing the efficiency of the process. The great success of CRISPR is mainly due to the simplicity and ease of gRNA design (only about 20 nucleotides are needed that will compose the guide RNA and will be complementary to the target region) [10]. In addition, it has a greater mutation efficiency and allows the manipulation of more than one gene at the same time, thus being able to generate multiple mutations [15, 16, 17], without the problem of off-target effects seen in TALENs and ZFNs [18, 19].
References
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T et al (2013) Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369:819–829
Krueger U, Bergauer T, Kaufmann B, Wolter I, Pilk S, Heider-Fabian M et al (2007) Insights into effective RNAi gained from large-scale siRNA validation screening. Oligonucleotides 17:237–250
Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S et al (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A et al (2014) Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510:235–240
Kim HJ, Lee HJ, Kim H, Cho SW, Kim J-S (2009) Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19:1279–1288
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S et al (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP et al (2011) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res 39:359–372
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Cho SW, Kim S, Kim JM, Kim J-S (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ et al (2013) A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31:251–258
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30:460–465
Li W, Teng F, Li T, Zhou Q (2013) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31:684–686
Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L et al (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156:836–843
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F et al (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK et al (2012) TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40:W117–W122
Şöllü C, Pars K, Cornu TI (2010) Autonomous zinc-finger nuclease pairs for targeted chromosomal deletion. Nucleic Acids
Lino CA, Harper JC, Carney JP, Timlin JA (2018) Delivering CRISPR: a review of the challenges and approaches. Drug Deliv Nov 25(1):1234–1257. https://. doi
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol Dec; 169(12):5429–5433
Mojica FJ, Juez G, Rodríguez-Valera F (1993) Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol Aug 9(3):613–621
Mojica FJM, DõÂez-VillasenÄor C, Soria E, Juez G (2000) Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36:244–246
van Soolingen D, de Haas PE, Hermans PW et al (1993) Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J Clin Microbiol 31:1987–1995
Jansen R, Embden JD, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol Mar 43(6):1565–1575
Bolotin A, Quinquis B, Sorokin A, Dusko Ehrlich S (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561
Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol Feb; 60(2):174–182
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA and provide additional tools for evolutionary studies. Microbiology 151:653–663
Brouns SJ, Jore MM, Lundgren M et al (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964
Sampson TR, Weiss DS (2014) CRISPR-Cas systems: new players in gene regulation and bacterial physiology. Front Cell Infect Microbiol 4:37. https://doi.org/10.3389/fcimb.2014.00037
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P et al (2011) Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol Jun 9(6):467–477
Barrangou R, Horvath P (2012) CRISPR: new horizons in phage resistance and strain identification. Annu Rev Food Sci Technol 3:143–162
Westra ER, Buckling A, Fineran PC (2014) CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol 12:317–326
Barrangou R, Marraffini LA (2014) CRISPR-Cas systems: prokaryotes upgrade to adaptative immunity. Mol Cell 54:234–244
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Jiang W, Marraffini LA (2015) CRISPR-Cas: New Tools for Genetic Manipulations from Bacterial Immunity Systems. Annu Rev Microbiol 69:209–228
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S et al (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31:833–838
Hatoum-Aslan A, Marraffini LA (2014) Impact of CRISPR immunity on the emergence and virulence of bacterial pathogens. Curr Opin Microbiol 0:8290. https://doi.org/10.1016/j.mib.2013.12.001
Chylinski K, Le Rhun A, Charpentier E (2013) The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol 10:726–737
Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y et al (2016) Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell May 165(4):949–962
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier EA (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science Aug; 337(6096):816–821
Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096–1258099
Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol Feb 190(4):1390–1400. doi: https://doi.org/10.1128/JB.01412-07. Epub 2007 Dec 7. PMID: 18065545; PMCID: PMC2238228
Garneau JE, Dupuis M, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. Nov 4;468(7320):67–71. doi: https://doi.org/10.1038/nature09523. PMID: 21048762
Jiang F, Doudna JA (2017) CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys May 22:46:505–529. https://doi.org/10.1146/annurev-biophys-062215-010822
Anders C, Niewoehner O, Duerst A, Jinek M (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. Sep 25;513(7519):569 – 73. doi: https://doi.org/10.1038/nature13579. Epub 2014 Jul 27. PMID: 25079318; PMCID: PMC4176945
Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, Severinov K (2011) Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A Jun 21(25):10098–10103. doi: https://doi.org/10.1073/pnas.1104144108. Epub 2011 Jun 6. PMID: 21646539; PMCID: PMC3121866
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A Sep 25(39):E2579–E2586. doi: https://doi.org/10.1073/pnas.1208507109. Epub 2012 Sep 4. PMID: 22949671; PMCID: PMC3465414
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res Nov; 39(21):9275–9282. doi: https://doi.org/10.1093/nar/gkr606. Epub 2011 Aug 3. PMID: 21813460; PMCID: PMC3241640
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015 Apr 9;520(7546):186 – 91. doi: https://doi.org/10.1038/nature14299. PMID: 25830891; PMCID: PMC4393360
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163(3):759–771. https://doi.org/10.1016/j.cell.2015.09.038
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F (2017) Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35(1):31–34. doi: https://doi.org/10.1038/nbt.3737
Zhang Yu, Li CLongH, McAnally JR, Kedryn K, Baskin JM, Shelton, Olson (2017) CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Science Advances, 2017; 3 (4): e1602814 DOI: https://doi.org/10.1126/sciadv.1602814
Strachan T, Read A (2013) Genética molecular humana. 4.ed. Artmed
Zhang C, Xiao B, Jiang Y, Zhao Y, Li Z, Gao H et al (2014) Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. MBio Jul; 5(4):e01414–e01414
Wang JY, Hoel CM, Al-Shayeb B et al (2021) ) Coordenação estrutural entre sítios ativos de um complexo CRISPR transcriptase reversa-integrase. Nat Commun 12:2571. https://doi.org/10.1038/s41467-021-22900-y
Liu JJ, Orlova N, Oakes BL et al (2019) ) CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 566:218–223. https://doi.org/10.1038/s41586-019-0908-x
Pausch P, Al-Shayeb B, Bisom-Rapp E, Tsuchida CA, Li Z, Cress BF, Knott GJ, Jacobsen SE, Banfield JF, Doudna J (2020) CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369(6501):333–337. doi: 10.1126/science.abb1400. PMID: 32675376; PMCID: PMC8207990
Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, Koonin EV, Zhang F (2017) Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol Cell 65(4):618–630e7. doi: https://doi.org/10.1016/j.molcel.2016.12.023
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, Lander ES, Voytas DF, Ting AY, Zhang F (2017) RNA targeting with CRISPR-Cas13. Nature 550(7675):280–284. doi: 10.1038/nature24049. PMID: 28976959; PMCID: PMC5706658
Oliver Smithies RGG, Boggs SS, Koralewski MA (1985) Insertion of DNA sequences into the human chromosomal b-globin locus by homologous recombination. Nature 317:19
Bibikova M, Golic M, Golic K, Carroll GD (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161:1169–1175
Christian M, Cermak T, Doyle EL et al (2010) Targeting DNA doublestrand breaks with TAL effector nucleases. Genetics 186:757–761
Funding
The authors are grateful to the Foundation for Science and Technology (FCT, Portugal) and FEDER under Programme PT2020 for financial support to CIMO (UID/AGR/00690/2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author Patrick Ferreira declares that he has no conflict of interest.
Author Altino Choupina declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ferreira, P., Choupina, A.B. CRISPR/Cas9 a simple, inexpensive and effective technique for gene editing. Mol Biol Rep 49, 7079–7086 (2022). https://doi.org/10.1007/s11033-022-07442-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07442-w