
Abstract
Topoisomerase IIβ binding protein 1 (TopBP1) is involved in cell survival, DNA replication, DNA damage repair and cell cycle checkpoint control. The biological function of TopBP1 and its close relation with BRCA1 prompted us to investigate whether alterations in the TopBP1 gene can influence the risk of breast cancer. The aim of this study was to examine the association between five polymorphisms (rs185903567, rs116645643, rs115160714, rs116195487, and rs112843513) located in the 3′UTR region of the TopBP1 gene and breast cancer risk as well as allele-specific gene expression. Five hundred thirty-four breast cancer patients and 556 population controls were genotyped for these SNPs. Allele-specific TopBP1 mRNA and protein expressions were determined by using real time PCR and western blotting methods, respectively. Only one SNP (rs115160714) showed an association with breast cancer. Compared to homozygous common allele carriers, heterozygous and homozygous for the T variant had significantly increased risk of breast cancer (adjusted odds ratio = 3.81, 95 % confidence interval: 1.63–8.34, p = 0.001). Mean TopBP1 mRNA and protein expression were higher in the individuals with the CT or TT genotype. There was a significant association between the rs115160714 and tumor grade and stage. Most carriers of minor allele had a high grade (G3) tumors classified as T2-T4N1M0. Our study raises a possibility that a genetic variation of TopBP1 may be implicated in the etiology of breast cancer.
Similar content being viewed by others

Introduction
Breast cancer is the most frequently diagnosed cancer and one of the leading causes of cancer death among women worldwide [1]. In Poland breast cancer is the second most common cause of cancer death in women. Moreover, breast cancer incidence rates have been reported to increase by up to 5 % per year in developing countries [2]. Although environmental factors and lifestyle could contribute to the increased breast cancer risk, genetic factors are also implicated in the pathogenesis of the disease. Recent genome-wide and candidate gene association studies have identified some low-penetrance variants associated with breast cancer [3, 4]. However, despite great progress in the breast cancer studies the molecular mechanisms that contribute to breast carcinogenesis remain poorly understood. Thus, there is a necessity to identify all breast cancer susceptibility genes.
TopBP1 protein was first identified as an interacting partner for topoisomerase IIβ. TopBP1 gene comprising 28 exons is located on chromosome 3q22.1 and encodes a 1,522 amino acid protein (180 kDa) [5]. TopBP1 protein seems to be essential for maintenance of chromosomal integrity and cell proliferation. This protein appears to be involved in DNA replication, DNA damage response and cell cycle checkpoint control [6, 7]. The most striking feature of TopBP1 is that it has eight BRCA1 C-terminal (BRCT) domains which were first identified in breast cancer gene 1 (BRCA1) [8, 9]. BRCT domains, about 90 amino acids in length are hydrophobic and are involved in interaction with other proteins as well as in interaction with single and double-stranded DNA [10]. The C-terminal region of TopBP1 containing two BRCTs is responsible for interaction with topoisomerase and shows considerable similarities with BRCA1. Apart from structural similarities TopBP1 shares many other common features with BRCA1. Both TopBP1 and BRCA1 are strongly induced during S phase. Following ionizing radiation, TopBP1 is recruited to DNA breaks and colocalizes with Nbs1 (Nijmegen breakage syndrome 1), BRCA1 and 53BP1 (p53-binding protein 1) in nuclear foci. TopBP1 and BRCA1 also colocalize with proliferating cell nuclear antigen at stalled replication forks after a replication block. Both proteins are phosphorylated by ATM (ataxia telangiectasia mutated) kinase in response to DNA damage and DNA replication stress [5]. Moreover, it was shown that TopBP1 is involved in regulation of p53 activities during normal growth.
The biological function of TopBP1 and its close relation with BRCA1 prompted us to investigate whether genetic alterations in the TopBP1 gene can influence the risk of breast cancer. In present study we tested the effect of SNPs potentially located in the 3′UTR (3′untranslated region) region of the TopBP1 gene and listed in NCBI’s (National Center for Biotechnology Information) SNP database on breast cancer risk as well as on allele-specific mRNA/protein expression. There are five such SNPs—rs185903567 (G/A), rs116645643 (A/G), rs115160714 (C/T), rs116195487 (C/G), and rs112843513 (C/delC)]. We correlated obtained results with clinicopathological characteristics.
Materials and methods
Study population
This study involved 534 women with non-hereditary infiltrating ductal breast carcinomas (age range 43–81, mean age 54.76 ± 7.35) recruited between May 2003 and November 2010. The patients had a confirmed diagnosis of ductal breast cancer based on histopathological evaluation and were under treatment at the Polish Mothers Memorial Hospital Research Institute, Łódź, Poland. None of the recruited patients received preoperative chemo- or radiotherapy. Patients diagnosed with previous breast tumors or with tumors located elsewhere were excluded.
A group of 556 healthy Polish individuals were collected from the hospital routine controls of health and used as control. They were non-related women, that have never been diagnosed with breast tumors, other tumors or chronic disease and were randomly selected and frequency matched to the cases on age (age range 34–83, mean age 51.27 ± 11.18).
Blood samples were collected from all women participating in the study and additionally breast cancer tissues were obtained from patients with breast neoplasms. We enrolled only women born and living in central Poland (Łódź region). Informed consent was obtained from patients and controls, and ethics approval was obtained from the ethics commission of the Polish Mother’s Memorial Hospital, Research Institute (G4/2011).
Lifestyle risk factors
Study participants were interviewed using questionnaire that included socio-demographic, medical history, health related information, alcohol intake, smoking status, menstrual and reproductive histories, and exogenous hormone use. Medical records of patients were thoroughly reviewed. The tumor stages were classified according to the 1997 TNM staging system of the American Joint Committee on Cancer [11]. Tumors were graded according to the Bloom and Richardson classification, modified by Elston and Ellis [12]. A positive family history of breast cancer was defined as reporting of breast cancer in one or more first degree relatives. Body mass index (BMI) was calculated based on current weight in kilograms divided by height in meters squared.
The subjects were classified as never/rare drinkers, ex-drinkers, or current drinkers who consumed 1–8.9 U/week (light drinkers), 9–17.9 U/week (moderate drinkers), or 18 U/week (heavy drinkers), where 1 U = 22 g ethanol [13].
According to smoking status patients and controls were grouped into ‘‘never’’, “former” and “current” based on self-reported usage. Participants who reported smoking at least 100 cigarettes in their lifetime and who, at the time of survey, smoked either every day or some days were defined as Current Smoker. Participants who reported smoking at least 100 cigarettes in their lifetime and who, at the time of the survey, did not smoke at all were defined as Former Smoker. Participants who reported never having smoked 100 cigarettes were defined as Never Smoker.
Menopause was defined as the time of the last menstrual period (or menstrual flow of any amount). None of the women involved in the study had undergone a hysterectomy. Regular drug usage was defined as self-report use of oral contraceptives or menopausal hormones for 6 months or longer.
Blood sample collection, SNP selection and genotyping
Each genomic DNA sample was extracted from peripheral blood using FlexiGene® DNA Kit (Qiagen GmbH, Hilden, Germany). DNA concentration was determined by spectrophotometry. The single nucleotide polymorphisms (SNPs) rs185903567 (G/A), rs116645643 (A/G), rs115160714 (C/T), rs116195487 (C/G), rs112843513 (C/delC) located at the 3′UTR of TopBP1 gene and listed in the NCBI’s SNP database were evaluated. These polymorphisms were analyzed by PCR amplification and direct sequencing. The amplified region included the entire 3′UTR region (nucleotides 4629-5289; NCBI Reference Sequence: NM_007027.3). Briefly, amplification was carried out in a final volume of 25 μl containing 100 ng genomic DNA, 0.3 μM of forward (5′-TGGGACTGGATTATCACAAAAG-3′) and reverse (5′-CTTTTATTCTTTATTGTCACATTTTCC-3′) primers, 0.2 mM dNTPs (deoxyribonucleoside triphosphates), 2 mM MgCl2, 1 × Buffer (Applied Biosystems, Darmstadt, Germany) and 1 U AmpliTaq Gold (Applied Biosystems, Darmstadt, Germany). PCR conditions were 94 °C for 5 min; 35 cycles with denaturation at 94 °C for 30 s, annealing at 61 °C for 30 s, and elongation at 72 °C for 30 s; and a final extension at 72 °C for 10 min. Purified PCR products were then sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Darmstadt, Germany) and electrophoresed on a 3730 DNA Analyzer (Applied Biosystems, Foster City, CA).
Total RNA extraction and cDNA synthesis
Total RNA was extracted from breast cancer tissues using TRI Reagent® (Sigma Aldrich Corp. St. Louis, MO, USA) according to manufacturer’s protocol. RNA was eluted in 20 μl RNase-free water, quantified by spectrophotometry at 260 nm and stored at −20 °C. RNA with a 260/280 nm ratio in range 1.8–2.0 was considered high quality. First-strand cDNAs were obtained by reverse transcription of 1 μg of total RNA using RevertAid™ First Strand cDNA Synthesis Kit (Fermentas UAB, Vilnius, Lithuania) following the manufacturer’s protocol.
Real time quantitative PCR
For real-time PCR analysis of TopBP1 mRNA in normal and pathological tissues, TaqMan® Gene Expression Assays (Applied Biosystems, Bedford, MA, USA) were used according to the manufacturer’s instruction. Before starting the real-time PCR analysis we used the NormFinder algorithm to select the best reference gene (http://www.mdl.dk). We chose GAPDH (glyceraldehyde 3-phosphate dehydrogenase) gene because it had the lowest stability value−0.017. The fluorogenic, FAM labeled probes and the sequence specific primers for TopBP1 and GAPDH were obtained as inventoried assays Hs00199775_m1 and Hs99999905_m1, respectively (Applied Biosystems, Bedford, MA, USA). The reactions were performed in duplicate. A positive result was defined by a threshold cycle (Ct) value lower than 40 (the Ct value is determined by the number of cycles needed to exceed the background signal). Ct value of all positive results were lower than 30. Abundance of TopBP1 mRNA in studied material was quantified by the ΔCt method. ΔCt (Ct TopBP1 − Ct GAPDH ) values were recalculated into relative copy number values (number of copies of TopBP1 mRNA per 1,000 copies of GAPDH mRNA).
Western blotting analysis
Tissue homogenate was obtained from each sample in the presence of the serine protease inhibitor PMSF (phenylmethylsulfonyl fluoride) and 10 mM sodium molybdate. The protein content was estimated by modified Lowry method using bovine serum albumin as standard. Homogenate proteins (50 μg protein/lane) were resolved by 8 % SDS-PAGE and electroblotted onto Immobilon-P transfer membranes (Millipore, Bedford, MA, USA). The blots were incubated 1 h with rabbit polyclonal anti-TopBP1 (Abcam, Cambridge, UK) in a 1:1,000 dilution. After being washed three times with TBST (Tris buffered saline with Tween-20), the membranes were incubated 1 h with goat anti-rabbit antibodies conjugated with horseradish peroxidase (1:5,000 dilution). The membranes were again washed three times with TBST and incubated with peroxidase substrate solution (3,3′-diaminobenzidine—DAB). Gel-Pro® Analyzer software (Media Cybernetics Inc., Bethesda, MD, USA) was used for densitometry analysis of protein bands. The integrated optical density (IOD) of the bands, in a digitized picture, was measured.
Evaluation of estrogen receptor and progesterone receptor
Estrogen receptor (ER) and progesterone receptor (PR) status was determined by immunohistochemical method as part of the routine clinical practice. Using the immunohistochemical assay, tumors were classified as positive if more than 10 % of the cells showed nuclear staining for the receptor. This information was received together with the characteristics of clinical material.
Quality control
For quality control purposes, 10 % of samples were randomly selected, and sequence analysis performed, with 100 % concordance to the genotype. Laboratory personnel were unable to distinguish among case, control, and quality control samples.
Statistical data analysis
Genotype distributions were evaluated for agreement with Hardy–Weinberg equilibrium by the Chi-square test. Unconditional multiple logistic regression models were used to calculate odds ratios (ORs) and 95 % confidence intervals (CIs) for the association of genotype with breast cancer risk. Genotype data were analyzed with the homozygote of the common allele as the reference group. Variants of homozygotes and heterozygotes were combined to evaluate the dominant effect. For each SNP, trend tests were conducted by assigning the ordinal values 1, 2, and 3 to homozygous wild-type, heterozygous, and homozygous variant genotypes, respectively, and by adding these scores as a continuous variable in logistic regression model. All multivariate models were adjusted for age, family history, obesity, smoking status, parity, menopausal status, and use of contraceptive and menopausal hormones. Since levels of TopBP1 mRNA and protein expression in studied material specimens did not show normal distribution (Kolmogorov–Smirnov test) the non-parametrical statistical tests (Mann–Whitney U test, Kruskal–Wallis test with post hoc multiple comparisons, Chi square test or the Spearman rank correlation test) were applied. Reported p values were two-sided. Probabilities were considered significant whenever p-value was lower than 0.05. All analyses were completed using SAS software (version 9.0 SAS Institute, Cary, NC, USA).
Results
Characteristics of subjects
The distributions of sociodemographic characteristics, lifestyle risk factors and clinical characteristics of the patients are shown in Tables 1 and 2, respectively. All patients and healthy subjects were Caucasian. The cases were slightly older (mean age 54.76 ± 7.35 vs. 51.27 ± 11.18), were more likely to have an BMI equal or greater than 30 (39.9 vs. 28.9 %) and more likely to use contraceptives (estrogens and progestins) and menopausal hormones 64.4 vs. 50.1 % and 42.3 vs. 30.9 %, respectively, than controls. Moreover, both groups slightly differ in smoking status. More patients currently smoke (37.1 vs. 26.2 %) and fewer had never smoked (18.0 vs. 27.9 %,) than women in control group.
Genotypes and genotypic distribution in patients and control subjects
Genotype distributions for TopBP1 polymorphisms in 534 breast cancer patients and 556 control subjects are summarized in Table 3. Two SNPs (rs116195487 and rs185903567) were not observed in the 3′UTR region of TopBP1 gene in our studied groups. During the study, we have not identified any new mutations, not listed in the SNP databases. All cases and controls were common allele carriers. Only one SNP (rs115160714) showed an association with breast cancer. The frequency of individuals who carried (T) allele was significantly higher in cases group (3.1 %) than in controls group (0.8 %; p < 0.001). Compared to homozygous common allele carriers, heterozygous for the T variant were found to be at a significant 3.54-fold increased risk of breast cancer (95 % CI = 1.56–8.39; p = 0.002). The TT genotype even more increased breast cancer risk compared with those harboring the CC genotypes (OR = 5.40, 95 % CI = 0.63–46.64; p = 0.004). The comparison of combined genotypes is shown in Table 4. Most cases and controls showed only one SNP polymorphism. However, nineteen cases (3.5 %) were heterozygotes for rs115160714 and had a C deletion in rs112843513.
Association of rs115160714 with clinical and environmental parameters
Of the 534 breast cancer patients, 406 (76.0 %) had a low grade tumor (grades G1 and G2), and 128 (24.0 %) had a high grade tumor (G3). Most tumors, 389 (72.8 %) was classified as T1-2N0M0, and the remaining 145 (27.2 %) was T2-4N1M0 (Table 5). There was a significant association between CT and TT genotypes and tumor grade or stage. Most carriers of minor allele had a high grade tumors classified as T2-4N1M0 (Table 5).
The analysis of SNP polymorphism (rs115160714) in smokers and non-smokers groups showed that smoking is a significant breast cancer risk factor in case of T allele carriers (Table 6). There was no association between alcohol intake and breast cancer risk.
Association between TopBP1 genotypes and mRNA/protein expression in breast cancer tissue
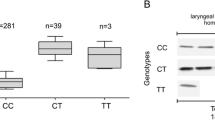
We found that mean TopBP1 mRNA expression was lower in the case of individuals with the CC genotype than in case of minor allele carriers, i.e. CT heterozygotes and TT homozygotes (223.0, 412.0 and 428.5 copies of TopBP1 mRNA per 1000 copies of GAPDH mRNA, respectively, p < 0.05 for all comparisons) (Table 7; Fig. 1a). We found TopBP1 protein expression in 81.2, 69.5 and 60.0 % of breast tissue homogenate samples of CC, CT, and TT genotype carriers, respectively. Although the protein expression was more frequently observed in common allele carriers group, the mean expression level was lower than in minor allele carriers (84.6, 118.2, 127.4 IOD relative units, respectively, p < 0.05 for all comparisons) (Table 7; Fig. 1b). There was a statistically significant correlation between TopBP1 mRNA and protein expressions (Spearman correlation coefficient for CC and CT genotype 0.76 and 0.82, respectively, p < 0.05 for all comparisons). However, not in all cases with positive mRNA expression we could detected TopBP1 protein. Both mRNA and protein was detected in 306 of 506 CC samples, in 14 of 23 of CT samples and 3 of 5 TT samples.

The relationship between TopBP1 mRNA and protein expression and the rs115160714 genotype in breast cancers. a Expression of TopBP1 gene measured by real-time PCR in relation to genotype. b Western blotting analysis of TopBP1 expression measured in relation to genotype. Figure shows the representative results of TopBP1 immunodetection in breast cancer tissue homogenates (50 μg protein per lane)
Discussion
The biological functions of TopBP1 protein as well as its close relation with BRCA1 suggest a crucial role of this protein in the maintenance of genome integrity and cell cycle regulation. Published data on the involvement of TopBP1 in breast carcinogenesis are very limited. However, the aberrant expression of TopBP1 protein in breast cancer was shown. Immunohistochemical analysis of TopBP1 level demonstrated that this protein was expressed almost exclusively in nuclei of the normal breast epithelium while in breast cancer samples TopBP1 was detected in nucleus and/or in cytoplasm [14]. Analysis of TopBP1 protein expression in feline and canine mammary neoplasms revealed that most TopBP1 immunohistochemical staining was nuclear but both nuclear and cytoplasmic staining was observed as the degree of malignancy increased. Expression of TopBP1 protein was also correlated with histological grade of neoplasms [14, 15]. Patients with overexpression of TopBP1 tend to have higher grades of breast cancer and negative estrogen receptor status compared with those without overexpression of this protein and have significantly shorter overall survival time [16]. The results of our earlier studies concerning expression of TopBP1 in hereditary breast cancer showed lower TopBP1 mRNA expression in lobular carcinoma compared with ductal carcinoma. The level of TopBP1 mRNA appeared to be lower in poorly differentiated (III grade) hereditary breast cancer in comparison with moderately (II grade) and well-differentiated cancer (I grade). However, the immunohistochemistry and Western blot analyzes showed significantly increased of TopBP1 protein level in poorly differentiated breast cancer (III grade). Our data suggested that increased level of TopBP1 protein might be associated with progression of hereditary breast cancer [17].
Two SNPs (rs116195487 and rs185903567) of the five listed in the NCBI’s SNP database were not observed in the 3′UTR region of TopBP1 gene in our studied groups. This allows to conclude that these polymorphisms do not occur in the Polish population.
In this study, we demonstrated for the first time that rs115160714 in the 3′UTR region of TopBP1 gene is significantly associated with breast cancer risk. Compared to homozygous common allele carriers, heterozygous for the T variant were found to be at a significant 3.54-fold increased risk of developing breast cancer (95 % CI = 1.56–8.39; p = 0.002).
Since genetic alteration to the 3′UTR sequence can increase or decrease the half- life of the mRNA leading to greater or lesser protein levels we presumed that rs115160714 may affect TopBP1 expression. We found out that mean TopBP1 mRNA and protein levels were higher in case of individuals with CT or TT genotype.
There are several regulatory sequences in the 3′UTR that can affect expression, i.e. polyadenylation signal, AU-rich elements and binding sites for miRNAs [18]. The rs115160714 was not located in or close to any of these sites except miRNAs binding sites. Thus we suggested that polymorphism rs115160714 could change stability of TopBP1 mRNA by affecting miRNA binding.
MicroRNAs are a class of regulatory RNAs reported to modulate various biological processes and predicted to regulate as many as 30 % of human mRNAs. The miRNA targeting is determined by the nature and extent of the complementarity between an miRNA and its target sequence in the 3′UTR of mRNA. Thus, a noncoding polymorphism residing in the miRNA or the miRNA target sequence may play a role in mRNA degradation or translational repression, which post-transcriptionally regulates gene expression, with a concomitant alteration in phenotype [19, 20].
To identify miRNAs that likely target the vicinity of the rs115160714 polymorphism in the TopBP1 3′UTR, we utilized a computational algorithm, MicoInspector (http://mirna.imbb.forth.gr/microinspector/) that yielded three candidate miRNAs, miR-3138, miR-4302 and miR-1207-5p, whose seed sequences are complementary to the TopBP1 mRNA sequence around the rs115160714 polymorphism.
We did not find any literature data about the miR-3138 and miR-4302 and there is only one study concerning miR-1207-5p. Papagregoriou et al. [21] demonstrated that variant 1936T of miRSNP (rs13385) in heparin binding epidermal growth factor (HBEGF) prevents miR-1207-5p from down-regulation of HBEGF in podocytes [22]. We speculate that our findings may be explained on the basis of a similar mechanism. Figure 2 shows a hypothetical diagram of the interaction between fragment of TopBP1 3′UTR sequence and miR-1207-5p. Nonetheless, further experiments with other algorithms are needed to prove above speculation.

Hypothetical depicting as rs115160714 (black cytosine inside a rectangle) in 3′UTR of TopBP1 is predicted to be targeted by miR-1207-5p. This base pairing is surrounded by the rectangle. miR-1207-5p sequence consists of white letters, while the letters in TopBP1 sequence are black. Sequences were identified with MicroInspector algorithm (http://mirna.imbb.forth.gr/microinspector/, changed)
We don’t know the exact biological consequences of changes in TopBP1 expression level. However, increased expression of TopBP1 can cause deregulation of p53 activity. TopBP1 interacts with p53 binding domain and inhibits the promoter-binding activity of p53 [16]. Alteration of balance between TopBP1 and p53 activity may have impact on breast carcinogenesis.
TopBP1 is an essential protein that has numerous roles in the maintenance of genomic integrity. In particular, it is required for the activation of ATR and participates in DNA damage response and DNA replication [9]. Taking into account the role of TopBP1 in DNA damage response we were interested in exploring whether TopBP1 SNP–breast cancer association varied according to smoking status or alcohol consumption. Tobacco smoking is the best recognized and most important risk factor of the development of malignant cancer. Tobacco smoke contains several potent chemical carcinogens and reactive oxygen species that may produce bulky adducts, oxidative DNA damage, and DNA strand breaks [23].
However, previous epidemiologic studies investigating the association of cigarette smoking and breast cancer showed inverse, null or positive associations. It has been suggested that the genetic background might modify the association between tobacco smoke and breast cancer. A few studies have shown that defective DNA repair system modestly increases tobacco-related breast cancer risk [24–28].
We found out significant breast cancer prevalence in group of smokers who were T allele carriers. Thus, polymorphisms in TopBP1 gene may modify the relationship between breast cancer and smoking.
There is strong epidemiological evidence that consumption of alcoholic beverages increases the risk of cancers of the oral cavity and pharynx, esophagus, and larynx. Alcohol drinking has also been linked to breast cancer in women [29]. Acetaldehyde, the primary metabolite of ethanol generates several types of DNA adducts that block DNA replication and affect DNA damage response [30]. Our results did not show any association between alcohol intake and increased risk of breast cancer in Polish population. TopBP1 polymorphism did not changed alcohol consumption-breast cancer risk relationship.
In conclusion, our results showed that rs115160714 polymorphism can increase breast cancer risk and is associated with changes in TopBP1 expression.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Coughlin SS, Ekwueme DU (2009) Breast cancer as a global health concern. Cancer Epidemiol 33:315–318
Peng S, Lü B, Ruan W, Zhu Y, Sheng H, Lai M (2011) Genetic polymorphisms and breast cancer risk: evidence from meta-analyses, pooled analyses, and genome-wide association studies. Breast Cancer Res Treat 127:309–324
Fanale D, Amodeo V, Corsini LR, Rizzo S, Bazan V, Russo A (2012) Breast cancer genome-wide association studies: there is strength in numbers. Oncogene 31:2121–2128. doi:10.1038/onc.2011.408
Forma E, Brys M, Krajewska W (2011) TopBP1 in DNA damage response. In: Kruman I (ed) DNA repair/book 4. INTECH Open Access Publisher, Rijeka, pp 281–304
Jeon Y, Ko E, Lee KY, Ko MJ, Park SY, Kang J, Jeon CH, Lee H, Hwang DS (2011) TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. J Biol Chem 286:5414–5422
Xu Y, Leffak M (2010) ATRIP from TopBP1 to ATR—in vitro activation of a DNA damage checkpoint. Proc Natl Acad Sci USA 107:13561–13562
Glover JNM (2006) Insights into the molecular basis of human hereditary breast cancer from studies of the BRCA1 BRCT domain. Fam Cancer 5:89–93
Sokka M, Parkkinen S, Pospiech H, Syväoja JE (2010) Function of TopBP1 in genome stability. Subcell Biochem 50:119–141
Rodriguez MC, Songyang Z (2008) BRCT domains: phosphopeptide binding and signaling modules. Front Biosci 13:5905–5915
Fleming ID, Cooper JS, Henson DE, Hutter R, Kennedy B, Murphy G, O’Sullivan B, Yarbo J, Sobin L (1997) AJCC cancer staging manual, 5th edn. Lippincott-Raven, Philadelphia
Elston CW, Ellis IO (1991) Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long term follow up. Histopathology 19:403–410
Yokoyama A, Kato H, Yokoyama T, Tsujinaka T, Muto M, Omori T, Haneda T, Kumagai Y, Igaki H, Yokoyama M, Watanabe H, Fukuda H, Yoshimizu H (2002) Genetic polymorphisms of alcohol and aldehyde dehydrogenases and glutathione S-transferase M1 and drinking, smoking, and diet in Japanese men with esophageal squamous cell carcinoma. Carcinogenesis 23:1851–1859
Morris JS, Nixon C, Bruck A, Nasir L, Morgan IM, Philbey AW (2008) Immunohistochemical expression of TopBP1 in feline mammary neoplasia in relation to histological grade, Ki67, ERalpha and p53. Vet J 175:218–226
Morris JS, Nixon C, King OJA, Morgan IM, Philbey AW (2009) Expression of TopBP1 in canine mammary neoplasia in relation to histological type, Ki67, ERα and p53. Vet J 179:422–429
Liu K, Bellam N, Lin HY, Wang B, Stockard CR, Grizzle WE, Lin WC (2009) Regulation of p53 by TopBP1: a potential mechanism for p53 inactivation in cancer. Mol Cell Biol 29:2673–2693
Forma E, Krześlak A, Bernaciak M, Romanowicz-Makowska H, Bryś M (2012) Expression of TopBP1 in hereditary breast cancer. Mol Biol Rep 39:7795–7804
Chatterjee S, Pal JK (2009) Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell 101:251–262
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Liu X, Fortin K, Mourelatos Z (2008) MicroRNAs: biogenesis and molecular functions. Brain Pathol 18:113–121
Papagregoriou G, Erguler K, Dweep H, Voskarides K, Koupepidou P, Athanasiou Y, Pierides A, Gretz N, Felekkis KN, Deltas C (2012) A miR-1207-5p binding site polymorphism abolishes regulation of HBEGF and is associated with disease severity in CFHR5 nephropathy. PLoS One 7:e31021
Karppinen SM, Erkko H, Reini K, Pospiech H, Heikkinen K, Rapakko K, Syväoja JE, Winqvist R (2006) Identification of a common polymorphism in the TopBP1 gene associated with hereditary susceptibility to breast and ovarian cancer. Eur J Cancer 42:2647–2652
Reynolds P, Hurley S, Goldberg DE, Anton-Culver H, Bernstein L, Deapen D, Horn-Ross PL, Peel D, Pinder R, Ross RK, West D, Wright WE, Ziogas A (2004) Active smoking, household passive smoking, and breast cancer: evidence from the California Teachers Study. J Natl Cancer Inst 96:29–37
Mechanic LE, Millikan RC, Player J, de Cotret AR, Winkel S, Worley K, Heard K, Heard K, Tse CK, Keku T (2006) Polymorphisms in nucleotide excision repair genes, smoking and breast cancer in African Americans and whites: a population-based case-control study. Carcinogenesis 27:1377–1385
Metsola K, Kataja V, Sillanpää P, Siivola P, Heikinheimo L, Eskelinen M, Kosma VM, Uusitupa M, Hirvonen A (2005) XRCC1 and XPD genetic polymorphisms, smoking and breast cancer risk in a Finnish case-control study. Breast Cancer Res 7:R987–R997
Pachkowski BF, Winkel S, Kubota Y, Swenberg JA, Millikan RC, Nakamura J (2006) XRCC1 genotype and breast cancer: functional studies and epidemiologic data show interactions between XRCC1 codon 280 His and smoking. Cancer Res 66:2860–2868
Shen J, Gammon MD, Terry MB, Wang L, Wang Q, Zhang F, Teitelbaum SL, Eng SM, Sagiv SK, Gaudet MM, Neugut AI, Santella RM (2005) Polymorphisms in XRCC1 modify the association between polycyclic aromatic hydrocarbon-DNA adducts, cigarette smoking, dietary antioxidants, and breast cancer risk. Cancer Epidemiol Biomarkers Prev 14:336–342
Shore RE, Zeleniuch-Jacquotte A, Currie D, Mohrenweiser H, Afanasyeva Y, Koenig KL, Arslan AA, Toniolo P, Wirgin I (2008) Polymorphisms in XPC and ERCC2 genes, smoking and breast cancer risk. Int J Cancer 122:2101–2105
Corrao G, Bagnardi V, Zambon A, La Vecchia C (2004) A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev Med 38:613–619
Brooks PJ, Theruvathu JA (2005) DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 35:187–193
Acknowledgments
This work was supported by the statutory fund for Department of Cytobiochemistry, University of Łódź.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ewa Forma and Ewa Brzeziańska contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Forma, E., Brzeziańska, E., Krześlak, A. et al. Association between the c.*229C>T polymorphism of the topoisomerase IIβ binding protein 1 (TopBP1) gene and breast cancer. Mol Biol Rep 40, 3493–3502 (2013). https://doi.org/10.1007/s11033-012-2424-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-012-2424-z