
Abstract
Induction and activation of the p53 tumour suppressor protein occurs in response to a number of cellular stresses, including disruption of RNA polymerase II-mediated transcription. Both p53 itself and its principle negative regulator, the E3 ubiquitin ligase Mdm2, are substrates for phosphorylation by the protein kinase CK2 in vitro. CK2 phosphorylates Mdm2 within its central acidic domain, a region that is critical for making a second point of contact with p53 and mediating p53 ubiquitylation and turnover. Additionally, there is evidence that CK2 interacts with, and regulates, both p53 and Mdm2 within the cell but the molecular mechanisms through which CK2-mediated regulation of the p53 response can occur are only poorly understood. Previously, we showed that the basal transcription factor TAFII250, a critical component of TFIID, can interact with Mdm2 and promote the association of the Mdm2 acidic domain with p53. In the present study, we show that immunoprecipitates of TAFII250, either from mammalian cell extracts or from baculovirus-infected cells expressing elevated levels of HA-tagged TAFII250, can phosphorylate Mdm2 in vitro within its acidic domain. We show that CK2 is tightly associated with TAFII250 and is the principal activity responsible for TAFII250-mediated phosphorylation of Mdm2. Our data fit with recent observations that phosphorylation of the acidic domain of Mdm2 stimulates its association with p53 and are consistent with a model in which recruitment of CK2 by TAFII250 may play a contributory role in the maintenance of Mdm2 phosphorylation and function.
Similar content being viewed by others


Introduction
The p53 tumour suppressor protein is a short-lived transcription factor that is stabilised and activated in response to a wide variety of cellular stresses, including DNA damage, hyper-proliferation, and disruption of RNA polymerase II-dependent transcription (reviewed in [1–3]). Activated p53 orchestrates the transactivation or transrepression of many target genes leading to growth arrest, senescence, or apoptosis. The oncoprotein Mdm2 is a critical regulator of p53 that mediates the ubiquitylation and rapid proteasome-dependent degradation of both p53 and Mdm2 itself [4, 5]; reviewed in [6]. Cellular stresses that induce p53 disrupt the p53-Mdm2 interaction with the effect of attenuating p53 degradation [7].
Mdm2 comprises several conserved regions including an N-terminal p53 binding domain, a C-terminal RING finger domain required to mediate ubiquitylation of p53 and auto-ubiquitylation [4, 5], and a central acidic region that is critical for p53 degradation [8–12]. Thus, while p53-Mdm2 association occurs initially between the N-termini of the two proteins, this interaction is thought to lead to a conformational change in Mdm2 that permits a second point of contact between the core domain (DNA binding domain) of p53 and the acidic domain of Mdm2 [13–16]. This model explains why loss of the acidic region of Mdm2 results in a failure of Mdm2 to efficiently ubiquitylate and degrade p53 (e.g. see Ref. [17]).
The ability of Mdm2 to modulate p53 levels is tightly regulated through a variety of protein/protein interactions and post-translational modifications in response to a range of cellular stresses and alterations in growth/survival status [6, 18]. The acidic domain of Mdm2 is also highly phosphorylated on a cluster of serine residues under normal, homeostatic conditions [12, 19]. Several protein kinases have been proposed to phosphorylate sites within this region in vitro and in cultured cells [20–24] (see Fig. 1a). These include protein kinase CK2 which can phosphorylate ser269 and (albeit weakly) ser260 [20–22]. Alanine substitution of several of these sites (including the CK2 site, ser269) weakens or abolishes the ability of Mdm2 to mediate p53 turnover, suggesting that phosphorylation of key sites plays an important role in the normal maintenance of p53 turnover by Mdm2 [12, 22]. Strikingly, this region undergoes hypophosphorylation following DNA damage, consistent with the model that stress-induced dephosphorylation of the acidic domain inactivates Mdm2 and thereby contributes to the induction of p53 [12, 23].

Phosphorylation of the acidic domain of Mdm2. (a) Schematic representation of the Mdm2 protein highlighting functional domains including the N-terminal (principal) p53 binding site (p53BD), nuclear localisation (NLS) and export (NES) sequences, the acidic region, the Zn finger and the RING finger (responsible for the transfer of ubiquitin to p53). The expanded region represents the amino acid sequence of a portion of the acidic domain (human Mdm2) highlighting the known or putative phosphorylation sites and (in bold) the protein kinases thought to be responsible for the modification of these residues. The diagram summarises the conclusions of several studies [12, 13, 19, 23, 24]. (b) Amino acid sequence alignment of a portion of the acidic domain of Mdm2 with a region of similarity in the transcription factor, TFIIA. Amino acid identities are highlighted by grey boxes and sites of known phosphorylation in TFIIA are indicated [37]
The TATA binding protein (TBP)-associated factor (TAF), TAFII250 (also known as CCG1 or TAF1 in humans), acts as a scaffold for the assembly of transcription factor TFIID (comprising TBP and a number of other TAFs) and encompasses several biochemical activities that facilitate transcriptional initiation (reviewed in [25]). TAFII250 has been reported to be a protein kinase in its own right and to contain two so-called atypical protein kinase domains (at the N- and C-termini, respectively) [26, 27], neither of which bears any strong resemblance to the well-conserved classical protein kinase primary structure defined by Hanks [28]. Interestingly, the ability of TAFII250 protein kinase to phosphorylate p53 can be inhibited using the protein kinase CK2-specific inhibitor, apigenin [29], suggesting that either the inhibitor has a broader specificity than for CK2 alone, or that TAFII250 may be able to mediate phosphorylation of its substrates through the recruitment of protein kinases such as CK2.
Blockage of RNA polymerase II transcription is thought to be a key factor in the induction of p53 by agents that elicit DNA damage [1]. Consistent with this idea, inactivation of TAFII250 temperature-sensitive mutants leads to the induction and activation of the p53 pathway [17, 30, 31] through a mechanism thought to be dependent upon the protein kinase, ATR [30]. Additionally, TAFII250 interacts directly with the RING finger domain of Mdm2 [32] and can promote stabilisation of Mdm2 by blocking its auto-ubiquitylation function [17]. We also find that TAFII250 promotes Mdm2-dependent ubiquitylation of p53 by stimulating the interaction between the acidic domain of Mdm2 and the core region of p53 [17]. Interestingly, others have shown that phosphorylation of the Mdm2 acidic domain greatly enhances its ability to associate with p53 [13]. Taken together, these findings raise the possibility that TAFII250 could promote p53-Mdm2 interaction by mediating, or contributing towards, phosphorylation of the Mdm2 acidic domain.
In the present study, we have examined this hypothesis and show that TAFII250 in human cell extracts, and immunopurified recombinant TAFII250, associates tightly with protein kinase CK2 and can phosphorylate the acidic domain of Mdm2 in vitro in a manner that is sensitive to the CK2 inhibitor, TBB. Our findings also suggest that TAFII250 and/or the TFIID complex can recruit CK2 in a manner that permits phosphorylation of a physiological substrate, thereby providing a further example supporting the model that “regulation” of CK2 can occur through its dynamic recruitment to specific cellular processes [33, 34].
Materials and methods
Cell culture and baculovirus infection
HeLa and U2OS cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum (FCS) and 100 units/ml penicillin/streptomycin at 37°C and 5% CO2 in a humidified atmosphere.
Sf9 insect cells were infected with recombinant baculovirus expressing HA-tagged human TAFII250 (a kind gift from Frank Sauer and Tobias Maile). The recombinant protein was immunopurified using the anti-HA antibody, 12CA5.
Recombinant GST-Mdm2 fusion proteins used in the study were expressed in the plasmid pGEX2T (GE Healthcare) and have been described previously [35, 36] and are shown schematically in Fig. 2b. Expression of these proteins in Escherichia coli and their affinity purification using glutathione-sepharose 4B beads were performed according to the manufacturer’s instructions (GE Healthcare).

The acidic domain of Mdm2 is phosphorylated by TAFII250 in vitro. (a) S. frugiperda cells were mock-infected, or infected with baculovirus encoding HA-tagged TAFII250. After cell lysis, the TAFII250 was immunopurified and incubated in the presence of GST-Mdm2 and [γ-32P]ATP. Phosphorylated proteins were resolved by SDS-PAGE and detected by auto-radiography. (b) Schematic representation of the previously reported GST-Mdm2 fusion proteins used in the protein kinase experiments [35, 36]. The numbers indicate the amino acids of (human) Mdm2 present in each of the fusion proteins. (c) Phosphorylation of full length Mdm2 and domains of Mdm2 (as GST-fusion proteins) by baculovirus-expressed recombinant human TAFII250 in vitro using the conditions described in panel A. (d) Phosphorylation of the GST-Mdm2 fusion protein, MP3 in vitro by various immunoprecipitates from HeLa cell extracts (as indicated in the panel) using the conditions described in panel A
Cell lysates and ion exchange chromatography
All procedures were carried out at 4°C. Cells were lysed in NP-40 lysis buffer (10 mM Tris–HCl pH 7.5, 0.1 mM EDTA, 150 mM NaCl, 1% (v/v) Ipegal, 0.1% (v/v) 2-mercaptoethanol) supplemented with protease inhibitors. After clearing by centrifugation, the extracts were loaded onto a Mono Q (H/R 5/5) ion exchange column equilibrated in column buffer (50 mM Tris pH 7.5, 0.1 mM EDTA, 0.1% (v/v) 2-mercaptoethanol and supplemented with protease inhibitors). After loading the protein, the column was washed extensively with column buffer and the proteins were eluted with a linear gradient of 0–0.6 M NaCl (fractions 1–30) in the column buffer immediately followed by a sharp gradient from 0.6 to 1.0 M NaCl (fractions 31–35). Fractions were assayed for casein kinase and Mdm2 kinase activities and were analysed by western blotting for the presence of CK2 and TAFII250.
Immunoprecipitations and antibodies
For immunoprecipitation, cells were lysed in NP-40 lysis buffer (10 mM Tris–HCl pH 7.5, 2 mM EDTA, 150 mM NaCl, 1% (v/v) Ipegal) supplemented with protease inhibitors. Alternatively, fractions from the ion exchange chromatography were used for immunoprecipitation. Equal amounts of lysate or column fraction were incubated with 2 μg of anti-TAFII250 (clone 6B3, Upstate) or anti-Mdm2 (SMP14, 4B2 and/or D12, Santa Cruz). Recombinant HA-tagged TAFII250 was immunoprecipitated using the anti-HA monoclonal antibody 12CA5 (obtained from Cancer Research UK). Next, protein G Sepharose beads were added and the suspension was rocked gently for 2 h at 4°C. The beads were washed three times with NP-40 buffer, and 25 μl of 2× SDS loading buffer was added. Immunoprecipitated proteins were analysed by western blotting using TAFII250- or Mdm2-specific antibodies.
Western blotting
Cell extracts were subjected to SDS-PAGE and western blotting using standard conditions. Detection of Mdm2, TAFII250 and HA was carried out using the antibodies described in the previous section. Appropriate horse-radish peroxidase-coupled secondary antibodies were obtained from DakoCytomation. Detection was carried out by chemiluminescence.
Protein kinase assays
Substrate proteins (either 2–5 μg of casein or recombinant GST-Mdm2) were incubated in a total volume of 20 μl at 30°C in 50 mM Tris, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 10 mM NaF, 10 mM 2-glycerophosphate and 20 μM [γ-32P]ATP (specific activity 12 Ci/mmol). Reactions were initiated by the addition of protein kinase and terminated by the addition of an equal volume of 2× SDS sample buffer. Phosphorylated proteins were resolved by SDS-PAGE and detected by auto-radiography.
Results and discussion
A region of transcription factor TFIIA that is phosphorylated by TAFII250 shares similarity with the acidic domain of Mdm2
In order to understand better the molecular signalling mechanisms that regulate Mdm2 function, we carried out a BLAST search using the acidic domain of human Mdm2 (amino acids 247–270). The rationale behind this analysis was to determine whether clues could be provided concerning possible signalling systems responsible for Mdm2 regulation by comparison with similar or related, but well-characterised, regulatory domains in other proteins. This analysis indicated that the transcription factor TFIIA contains a region of similarity to amino acids 247–270 of Mdm2 (Fig. 1). This region of TFIIA is known to be phosphorylated at two sites, serines 316 and 321. Remarkably, the positions of these residues match sites of phosphorylation of Mdm2 in our alignment (Fig. 1b), raising the possibility that the protein kinase(s) that phosphorylates TFIIA might also phosphorylate Mdm2. Strikingly, the TFIIA-Ser316/321 protein kinase has been identified as TAFII250 [37].
The acidic domain of Mdm2 is phosphorylated by TAFII250 in vitro
To determine whether TAFII250 could phosphorylate Mdm2 in vitro, purified recombinant GST-Mdm2 was incubated in the absence or presence of immunoprecipitated HA-tagged TAFII250 together with [γ-32P]ATP as phosphate donor. The data (Fig. 2a) show that phosphorylation of Mdm2 occurred when the TAFII250 was immunoprecipitated from Sf9 cells that had been infected with the TAFII250-encoding baculovirus but not from the mock-infected cells. When GST alone was used as a substrate, no incorporation of phosphate could be detected (Fig. 2c, lane 1 and data not shown). Similar results were obtained when TAFII250 immunoprecipitated from HeLa cell extracts using an anti-TAFII250 antibody was used as the source of protein kinase activity although a much weaker signal was obtained owing to lower levels of TAFII250 available (data not shown). To determine the location of the phosphorylation event(s) in the Mdm2 molecule, a series of GST-Mdm2 fusion proteins, comprising overlapping regions of the human Mdm2 protein (Fig. 2b; [35, 36]), was used as substrates for the immunoprecipitated HA-tagged TAFII250. These analyses clearly showed that the phosphorylation occurred solely with the GST-Mdm2 fusion protein termed MP3, comprising amino acids 203–282 and containing the acidic domain (Fig. 2c). Subsequent analyses, using immunoprecipitates of TAFII250 or TBP from HeLa cells extracts, confirmed the presence of MP3 protein kinase activity, either within the TAFII250 molecule itself or associated with the TFIID complex (Fig. 2d). In contrast, only very weak activity was observed in non-specific or actin immunoprecipitates (Fig. 2d).
TAFII250 phosphorylates Mdm2 mainly through its ability to recruit protein kinase CK2
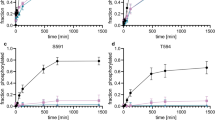
Having identified TAFII250 as a potential Mdm2 kinase, HeLa cell extracts were fractionated by ion exchange chromatography to determine whether TAFII250 might co-purify with any major detectable peak of Mdm2-directed kinase activity. Analysis of the fractions revealed two major peaks of protein kinase activity able to phosphorylate the acidic region of Mdm2 (using MP3 as substrate; Fig. 3a); these peaks coincided with the peaks to CK1 (fractions 13–15) and CK2 (fractions 27–28; Fig. 3a) and confirmed previous observations that both of these enzymes are highly active towards the acidic region of Mdm2 [20–22, 24]. Western blotting analysis confirmed that the second major peak contained protein kinase CK2 (Fig. 3a). Interestingly, TAFII250 eluted from this column coincident with CK2 (as detected by western blotting; Fig. 3a). Notably, the Mdm2 kinase activity present in this fraction was acutely sensitive to the CK2 inhibitor, TBB, with approximately 90% of the activity lost in the presence of 1 μM TBB (Fig. 3b). While this strongly suggested that CK2 was principally responsible for the phosphorylation of Mdm2, we cannot rule out the possibility that TAFII250 itself is inhibited by TBB, as has been reported for another CK2 inhibitor, apigenin [29]. These data also raised the possibility that TAFII250 could be associated with, and mediate its protein kinase function through, CK2. To explore the plausibility of this idea, the interaction between TAFII250 and CK2 was analysed by co-immunoprecipitation analysis using the peak fraction from the ion exchange chromatography containing the two proteins. Strikingly, immunoprecipitates of TAFII250 contained detectable CK2α, while in the reciprocal analysis, immunoprecipitates of CK2α also contained detectable TAFII250 (Fig. 3c). The observation that CK2α was present in immunoprecipitates of TAFII250 from U2OS cells confirmed that this interaction was neither cell line-specific nor an artefact of the fractionation procedure (Fig. 3d). Moreover, the observation that TBB is potently able to inhibit the ability of immunopurified TAFII250 to phosphorylate Mdm2 lends support to the idea that associated CK2 mediates phosphorylation of Mdm2 by TAFII250. However, the lack of complete inhibition, even at high levels of TBB (Fig. 3e), suggests that additional protein kinase activities may be associated with TAFII250.

Association between TAFII250 and protein kinase CK2. (a) HeLa cell extracts were loaded onto a Mono Q (H/R 5/5) column and the proteins eluted using a linear gradient of 0–600 mM NaCl (fractions 1–30). Fractions were assayed for the presence of Mdm2 kinase using the GST-Mdm2 fusion protein MP3, and for casein kinase using casein as substrate in the presence of [γ-32P]ATP (upper two panels, respectively). The presence of protein kinase CK2 and TAFII250 in the fractions was assessed by western blotting using appropriate antibodies (lower two panels). (b) Inhibition of Mdm2 (MP3) kinase activity in peak CK2 fraction (number 28) using increasing concentrations of the CK2-specific inhibitor, TBB. (c) Co-immunoprecipitation of TAFII250 and CK2 from Mono Q fraction 28. (d) Co-immunoprecipitation of TAFII250 and CK2 from extracts of U2OS cells transfected with a plasmid expressing untagged TAFII250. (e) Immunoprecipitated HA-tagged TAFII250 (HA) or mock immunoprecipitates (IgG) were used to phosphorylate the GST-Mdm2 fusion protein, MP3, in vitro in the absence or presence of 25 μM TBB
Other observations fit with the idea that it is the associated CK2 that is responsible for the ability of TAFII250 to phosphorylate Mdm2: (a) CK2 has been reported independently to associate with TBP (through the β subunit) [38, 39] and could, therefore, be an additional critical component of TFIID. (b) TAFII250 can phosphorylate thr55 of p53 in a manner that can be inhibited by the CK2 inhibitor, apigenin [29], again raising the possibility that CK2 provides (at least some of) the protein kinase activity attributed to TAFII250. (c) We have cloned and expressed both the NTK and CTK regions of TAFII250 in E. coli but have not been able to detect any trans- or auto-phosphorylation activities associated with these domains (data not shown); however, we cannot rule out the possibility that these domains may require other parts of the TAFII250 molecule to achieve detectable function, or may depend on interaction with, or modification by, other mammalian cellular proteins for appropriate folding or activation.
A model for the TAFII250-mediated regulation of p53 turnover
p53 and Mdm2 interact through a well-characterised and tight association involving the N-terminal regions of each protein. The binding of p53 to the N-terminus of Mdm2 is thought to give rise to a conformational change in Mdm2 that permits the acidic domain to make a second point of contact with p53 within its DNA binding domain, that is necessary for efficient ubiquitylation of p53 to occur [13–16]. The acidic domain is critical for this process because Mdm2 fails to ubiquitylate p53 efficiently in its absence [8–12, 17]. Recent evidence has suggested that the acidic domain, which is normally highly phosphorylated, makes a significantly tighter association with p53 in its phosphorylated as compared with its unphosphorylated state [13]. Our own recent data indicate that TAFII250 can stabilise Mdm2 and stimulate the association between p53 and the acidic domain of Mdm2, thereby permitting efficient ubiquitylation and degradation of p53 [17] (described schematically in Fig. 4) Based on the findings reported in the present manuscript, we propose that one plausible molecular mechanism through which TAFII250 can stimulate p53/Mdm2 contact would be through the recruitment of protein kinases, including CK2, that are known to phosphorylate the acidic domain. This model is based essentially on biochemical analyses and will need to be supported by subsequent molecular/cell-based studies. However, if the model is correct, we would expect the following predictions to hold true: (a) p53/Mdm2 contact, and indeed p53 ubiquitylation itself, should be sensitive to inhibitors of protein kinase CK2 or to siRNA-based knock-down of CK2 catalytic subunit; (b) inactivation of TAFII250 (e.g. by shifting the temperature-sensitive TAFII250 mutant to the non-permissive temperature) should lead to hypophosphorylation of the acidic domain of Mdm2.

Proposed model for the regulation of Mdm2 by TAFII250 and the involvement of protein kinase CK2. The model is discussed in depth in the text
In addition to the predictions proposed above, the model raises a number of new questions that should now be addressed. These are as follows: (a) given the principle that recruitment of CK2 into specific complexes can influence the substrate selectivity of the kinase (e.g. association with the transcriptional elongation factor, FACT, can preferentially direct CK2 activity towards the ser392 site of p53 [40]) does TAFII250-associated CK2 still preferentially phosphorylate ser269 of Mdm2 or is its activity re-directed to other or weaker (e.g. ser260 [20]) sites in the acidic domain? Our initial attempts to address this issue by chymo-tryptic phospho-peptide mapping have been inconclusive (data not shown) but may be improved with the development of new phospho-specific antibodies towards sites in the acidic domain. (b) Does TAFII250 recruit additional protein kinases to phosphorylate Mdm2? The inability to completely inhibit TAFII250-associated Mdm2 kinase activity suggests that either the TAFII250 molecule itself retains TBB-insensitive kinase activity or that other protein kinases may, like CK2, be associated with the TFIID complex. Either way, this raises the possibility that TAFII250 may act in the form of a signalosome that can direct protein kinase activity to key events in transcription. Such a notion would also be consistent with the growing perception that control of CK2 function can occur through its dynamic recruitment to specific cellular processes [33, 34]. (c) How does the TAFII250/CK2-mediated phosphorylation of Mdm2 fit into the physiological context of TAFII250 as a critical mediator of transcriptional initiation and as a central component of the TFIID complex? There is growing evidence of an important link between RNA polymerase II-dependent transcription and the p53 pathway such that p53 is induced when transcriptional integrity is impaired (reviewed and discussed in [1]). Thus, inactivation of TAFII250 not only disrupts transcription but also induces p53 [17, 30, 31]. The link between transcription and the maintenance of p53 levels is likely to be complex and clearly involves mechanisms such as regulation of Mdm2 expression and ATR-mediated activation of p53 [30, 31]. Additionally we have demonstrated a direct effect of TAFII250 on Mdm2 levels and on the p53-Mdm2 interaction [17]. Based on these observations it is possible that Mdm2 interacts with TAFII250 and/or other components of the transcriptional machinery and thus maintains p53 at physiological levels. This could occur, for example, on promoters themselves during the initiation stages of transcription. Disruption of transcriptional complexes could therefore eliminate direct communication with Mdm2 leading to a reduction in its activity and elevation of p53 levels. Based on the model presented above, phosphorylation of Mdm2 by CK2, as mediated through its association with TAFII250 (or other components of TFIID), may be a critical factor in this communication. Disruption of transcription by drugs, or by damage to the DNA itself, could interrupt this signalling mechanism leading to hypophosphorylation of Mdm2 and induction of p53.
Finally, this model could have relevance to tumour development. Protein kinase CK2 is found at abnormally high levels in a variety of cancers and over-expression of CK2α is tumorigenic in transgenic animals [41–45]. Elevated CK2 could help maintain Mdm2 in a highly active form making it more difficult for the cell to mount a p53 response. Transgenic animals expressing elevated levels of CK2α would provide a potential route to examine the effects of CK2 on p53 activation mechanisms. This approach, and analysis of the other issues discussed above, should reveal some interesting insights to the functional and pathological relationship between the p53 pathway, transcriptional integrity and the protein kinase CK2.
References
Ljungman M, Lane DP (2004) Transcription—guarding the genome by sensing DNA damage. Nat Rev Cancer 4:727–737. doi:10.1038/nrc1435
Vousden KH, Lu X (2002) Live or let die: the cell’s response to p53. Nat Rev Cancer 2:594–604. doi:10.1038/nrc864
Yee KS, Vousden KH (2005) Complicating the complexity of p53. Carcinogenesis 28:1317–1322. doi:10.1093/carcin/bgi122
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM (2000) Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275:8945–8951. doi:10.1074/jbc.275.12.8945
Honda R, Yasuda H (2000) Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19:1473–1476. doi:10.1038/sj.onc.1203464
Michael D, Oren M (2003) The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 13:49–58. doi:10.1016/S1044-579X(02)00099-8
Ashcroft M, Taya Y, Vousden KH (2000) Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 20:3224–3233. doi:10.1128/MCB.20.9.3224-3233.2000
Argentini M, Barboule N, Wasylyk B (2001) The contribution of the acidic domain of MDM2 to p53 and MDM2 stability. Oncogene 20:1267–1275. doi:10.1038/sj.onc.1204241
Kawai H, Wiederschain D, Yuan Z-M (2003) Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol Cell Biol 23:4939–4947. doi:10.1128/MCB.23.14.4939-4947.2003
Meulmeester E, Frenk R, Stad R, de Graaf P, Marine J-C, Vousden KH et al (2003) Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol 23:4929–4938. doi:10.1128/MCB.23.14.4929-4938.2003
Zhu Q, Yao J, Wani G, Wani MA, Wani AA (2001) Mdm2 mutant defective in binding p300 promotes ubiquitination but not degradation of p53: evidence for the role of p300 in integrating ubiquitination and proteolysis. J Biol Chem 276:29695–29701. doi:10.1074/jbc.M102634200
Blattner C, Hay TJ, Meek DW, Lane DP (2002) Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol 22:6170–6182. doi:10.1128/MCB.22.17.6170-6182.2002
Kulikov R, Winter M, Blattner C (2006) Binding of p53 to the central domain of Mdm2 is regulated by phosphorylation. J Biol Chem 281:28575–28583. doi:10.1074/jbc.M513311200
Shimizu H, Burch LR, Smith AJ, Dornan D, Wallace M, Ball KL et al (2002) The conformationally flexible S9–S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J Biol Chem 277:28446–28458. doi:10.1074/jbc.M202296200
Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL (2006) Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell 23:251–263. doi:10.1016/j.molcel.2006.05.029
Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR (2006) The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci USA 103:1227–1232. doi:10.1073/pnas.0510343103
Allende-Vega N, Saville MK, Meek DW (2007) Transcription factor TAFII250 promotes Mdm2-dependent turnover of p53. Oncogene 26:4234–4242. doi:10.1038/sj.onc.1210209
Meek DW, Knippschild U (2003) Post-translational modification of Mdm2. Mol Cancer Res 1:1017–1026
Hay TJ, Meek DW (2000) Multiple sites of in vivo phosphorylation in the MDM2 oncoprotein cluster within two important functional domains. FEBS Lett 478:183–186. doi:10.1016/S0014-5793(00)01850-0
Allende-Vega N, Dias S, Milne D, Meek D (2005) Phosphorylation of the acidic domain of Mdm2 by protein kinase CK2. Mol Cell Biochem 274:85–90. doi:10.1007/s11010-005-3074-4
Gotz C, Kartarius S, Scholtes P, Nastainczyk W, Montenarh M (1999) Identification of a CK2 phosphorylation site in mdm2. Eur J Biochem 266:493–501. doi:10.1046/j.1432-1327.1999.00882.x
Hjerrild M, Milne D, Dumaz N, Hay T, Issinger OG, Meek D (2001) Phosphorylation of murine double minute clone 2 (MDM2) protein at serine-267 by protein kinase CK2 in vitro and in cultured cells. Biochem J 355:347–356. doi:10.1042/0264-6021:3550347
Kulikov R, Boehme KA, Blattner C (2005) Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol 25:7170–7180. doi:10.1128/MCB.25.16.7170-7180.2005
Winter M, Milne D, Dias S, Kulikov R, Knippschild U, Blattner C et al (2004) Protein kinase CK1-delta phosphorylates key sites in the acidic domain of Mdm2 that regulate p53 turnover. Biochemistry 43:16356–16364. doi:10.1021/bi0489255
Wasserman DA, Sauer F (2001) TAFII250: a transcriptional toolbox. J Cell Sci 114:2895–2902
O’Brien T, Tjian R (1998) Functional analysis of the human TAFII250 N-terminal kinase domain. Mol Cell 1:905–911. doi:10.1016/S1097-2765(00)80089-1
Dikstein R, Ruppert S, Tjian R (1996) TAFII250 is a bipartite protein kinase that phosphorylates the base transcription factor RAP74. Cell 84:781–790. doi:10.1016/S0092-8674(00)81055-7
Hanks SK, Quinn AM, Hunter T (1988) The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241:42–52. doi:10.1126/science.3291115
Li HH, Li AG, Sheppard HM, Liu X (2004) Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol Cell 13:867–878. doi:10.1016/S1097-2765(04)00123-6
Buchmann AM, Skaar JR, DeCaprio JA (2004) Activation of a DNA damage checkpoint response in a TAF1-defective cell line. Mol Cell Biol 24:5332–5339. doi:10.1128/MCB.24.12.5332-5339.2004
Wasylyk C, Wasylyk B (2000) Defect in the p53-Mdm2 autoregulatory loop resulting from inactivation of TAF(II)250 in cell cycle mutant tsBN462 cells. Mol Cell Biol 20:5554–5570. doi:10.1128/MCB.20.15.5554-5570.2000
Leveillard T, Wasylyk B (1997) The Mdm2 C-terminal region binds TAFII250 and is required for Mdm2 regulation of the cyclin A promoter. J Biol Chem 272:30651–30661. doi:10.1074/jbc.272.49.30651
Filhol O, Martiel J-L, Cochet C (2004) Protein kinase CK2: a new view of an old molecular complex. EMBO Rep 5:351–355. doi:10.1038/sj.embor.7400115
Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15. doi:10.1042/BJ20021469
Burch L, Scott M, Pohler E, Meek D, Hupp T (2004) Phage-peptide display identifies the death-activated protein kinase family as a novel modifier of the p53-inducible gene products MDM2 and p21WAF1. J Mol Biol 337:115–128. doi:10.1016/j.jmb.2003.10.081
Kurki S, Peltonen K, Kiviharju T, Latonen L, Ojala P, Meek D et al (2004) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5:465–475. doi:10.1016/S1535-6108(04)00110-2
Solow S, Salunek M, Ryan R, LP M (2001) TAF(II)250 phosphorylates human transcription factor IIA on serine residues important for TBP binding and transcription activity. J Biol Chem 276:15886–15892. doi:10.1074/jbc.M009385200
Cabrejos ME, Allende CC, Maldonaldo E (2004) Effects of phosphorylation by protein kinase CK2 on the human basal components of the RNA polymerase II transcription machinery. J Cell Biochem 93:2–10. doi:10.1002/jcb.20209
Maldonado E, Allende JE (1999) Phosphorylation of yeast TBP by protein kinase CK2 reduces its specific binding to DNA. FEBS Lett 443:256–260. doi:10.1016/S0014-5793(98)01734-7
Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H et al (2001) A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell 7:283–292. doi:10.1016/S1097-2765(01)00176-9
Daya-Makin M, Sanghera JS, Mogentale TL, Lipp M, Parchomchuk J, Hogg JC et al (1994) Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res 54:2262–2268
Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K (1996) Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett 101:31–35. doi:10.1016/0304-3835(96)04110-9
Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC (2001) Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 20:3247–3257. doi:10.1038/sj.onc.1204411
Stalter G, Siemer S, Becht E, Ziegler M, Remberger K, Issinger OG (1994) Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem Biophys Res Commun 202:141–147. doi:10.1006/bbrc.1994.1904
Yenice S, Davis AT, Gouelli SA, Akdas A, Limas C, Ahmed K (1994) Nuclear casein kinase 2 (CK-2) activity in human normal, benign hyperplastic, and cancerous prostate. Prostate 24:11–16. doi:10.1002/pros.2990240105
Acknowledgements
We are grateful to Frank Sauer and Tobias Maile for providing us with baculovirus encoding HA-tagged human TAFII250. This study was supported by funding from the Association for International Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Allende-Vega, N., McKenzie, L. & Meek, D. Transcription factor TAFII250 phosphorylates the acidic domain of Mdm2 through recruitment of protein kinase CK2. Mol Cell Biochem 316, 99–106 (2008). https://doi.org/10.1007/s11010-008-9816-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9816-3