
Abstract
One of the most common gynecologic cancers is ovarian cancer and ranked third after the other two most common cancers: cervical and uterine. The highest mortality rate has been observed in the case of ovarian cancer. To treat ovarian cancer, an immune-informatics approach was used to design a multi-epitope vaccine (MEV) structure. Epitopes prediction of the cancer testis antigens (NY-ESO-1), A-Kinase anchor protein (AKAP4), Acrosin binding protein (ACRBP), Piwi-like protein (PIWIL3), and cancer testis antigen 2 (LAGE-1) was done. Non-toxic, highly antigenic, non-allergenic, and overlapping epitopes were shortlisted for vaccine construction. Chosen T-cell epitopes displayed a robust binding attraction with their corresponding Human Leukocyte Antigen (HLA) alleles demonstrated 97.59% of population coverage. The vaccine peptide was established by uniting three key constituents, comprising the 14 epitopes of CD8 + cytotoxic T lymphocytes (CTLs), 5 helper epitopes, and the adjuvant. For the generation of the effective response of CD4 + cells towards the T-helper cells, granulocyte–macrophage-colony-stimulating factor (GM-CSF) was applied. With the addition of adjuvants and linkers, the construct size was 547 amino acids. The developed MEV structure was predicted to be antigenic, non-toxic, non-allergenic, and firm in nature. I-tasser anticipated the 3D construction of MEV. Moreover, disulfide engineering further enhanced the stability of the final vaccine protein. In-silico cloning and vaccine codon optimization were done to analyze the up-regulation of its expression. The outcomes established the vaccine’s immunogenicity and safety profile, besides its aptitude to encourage both humoral and cellular immune responses. The offered vaccine, grounded on our in-silico investigation, may be considered for ovarian cancer immunotherapy.
Similar content being viewed by others


Introduction
Cancer is the most prevalent cause of mortality in the world, and it is the most common hindrance in most countries to achieving the desired life expectancy. Ovarian cancer is one of the most common types of cancer in women. (Guleria et al. 2020). Ovarian cancer is an extremely lethal and nastiest prognosis ailment. The inability of early diagnosis and nearly non-specific symptoms, common ovarian cancers are identified at the III/IV stage. Five major histological forms of ovarian cancer can be identified, and most of them are epithelial (Edwards et al. 2016; Torre et al. 2018; Siegel and Miller 2016). Endometrioid (13%), mucinous (13%), and clear cell cancers (6%) are the three most prevalent types of ovarian cancer (Coburn et al. 2017). Benign ovarian tumors, as recommended, are threat indicators and precursors of epithelial ovarian cancer because ovarian cancer and benign ovarian tumors share equivalent etiological risk factors (Jordan SJ, Green AC, Whiteman DC, Webb PM, Study AC 2007). It has also been proposed that the fallopian tube may also contribute to the origination of ovarian cancer as from the distal ends of fallopian tube predecessor lesions on ovaries that could progress into the severe ovarian tumor (Kurman 2016). Clear cells and endometrioid tumors are well-thought-out to be caused by establishing endometrium on ovaries throughout retrograde menstruation (Kurman 2016). Due to its heterogeneous nature, the development and origin of mucinous cancer have not been well established (Guleria et al. 2020).
Although ovarian cancer has a lower occurrence than breast cancer, it is considered three times more lethal than breast cancer. By the year 2020, the mortality rate of ovarian cancer is considered to be raised significantly. There are different reasons for predicting a greater mortality rate by ovarian cancer, like the non-symptomatic and stealthy progression of malignant tumorous cells, slow progression of disease symptoms, and lastly, lacking the specific methods for diagnosis of cancer even at advanced stages. That is why this cancer is given the name ‘Silent killer’ (Henriksen et al. 2020; Danilova et al. 2020; Yousefi et al. 2017). Ovarian cancer (OC) has caused an estimated 239,000 new cases and 153,000 deaths annually around the globe. The highest rates for this ratio are 11.4 per 100,000 and 6.0 per 100,000 correspondingly and have been observed in Central and Eastern Europe. A low incidence rate has been observed in China, i.e., 4.1 per 100,000, compared to 21,290 cases and 14,180 deaths related to ovarian cancer in the USA in 2015. In contrast, there have been estimated about 52,100 fresh cases and 22,500 expiries in the same year in China (Yousefi et al. 2017).
The treatment is far from ideal; surgery, radiotherapy, and chemotherapy are conventional treatment strategies used so far; however, these options have significant limitations and are not effective in numerous cases (Henriksen et al. 2020). In the current eras, multiple immunotherapy techniques have enabled an advance in oncology, including recombinant cytokines, immune checkpoint inhibitors T-cell-mediated adaptive therapies, and dendritic cells (DC)-based vaccines (Danilova et al. 2020; Yousefi et al. 2017). Cancer/testicular antigens (CTA) is over-expressed in tumor expressing conditions but not in the case of normal healthy cells that’s why these are preferred targets among TTA. (Aguilera et al. 2011; Tio et al. 2019). It has been reported that different patients show a different profile and level of CTA expression with the same diagnosis (Seledtsov and Goncharov 2015; Li et al. 2002; Garcia-Soto et al. 2017,2014; Faramarzi and Ghafouri-Fard 2017; Kulkarni and Uversky VNJIjoms. 2017). Generally, there are two groups of CTAs; non- X CTAs coded by the Y chromosome and autosomes and CT-X antigens present on the X chromosome (Tio et al. 2019). It has been reported in recent clinical trials that these cancer vaccines showed promising benefits (Melero et al. 2014). Besides aiding as potential targets for cancer vaccines, CTAs can also play their part in developing cancerous cells and survival (Anderson et al. 1984; Gjerstorff et al. 2015).
A remarkable advancement in developmental techniques has been observed using bioinformatics tools and immuno-informatics in the twenty-first century. The degree of viral vaccine progress has been elevated by reverse and structural vaccinology (Calis et al. 2013). Using these tools and techniques, the antigenicity of protein can be anticipated easily with a significant amount of accuracy (Dimitrov et al. 2017). Different kinds of antigenic determinants and adjuvants are selected to increase the efficiency of the preparation of subunit vaccines. It helps to improve the immune system and host immune responses (Gupta et al. 2013). The use of immuno-informatics has recently been intensified to model peptide vaccines against viruses (Shahid et al. 2020; Tahir ul Qamar M, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F,, et al. 2002; Khalid and Ashfaq 2020; MT ul Qamar,, et al. 2020; MT ul Qamar, Rehman A, Ashfaq UA, Awan MQ, Fatima I, Shahid F,, et al. 2020), parasites (Yadav et al. 2020; Kar and Srivastava 2018), and bacteria (Nezafat et al. 2017; Hajighahramani et al. 2017; Mamede et al. 2008; Mahmood et al. 2021).
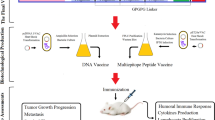
Bioinformatics progressions and technologies have become a new track for predicting the most vulnerable epitopes or designing MEV, i.e., a Multi-epitope-based subunit vaccine can trigger the humeral and cellular-based immune responses in the body (Saha 2006; Altschul et al. 1990). Herein, a multi-epitope subunit vaccine was designed to treat ovarian cancer. The vaccine was planned and constructed by predicting the epitopes of targeted cancer testes antigens such as NY-ESO-1, AKAP4, ACRBP, SPAGE9, MAGEA9, CT45A3, PIWIL3, LAGE-1. GM-CSF is used as an immune modulator was also utilized. For augmenting a cell-mediated and antibody-mediated immunity, GM-CSF was also incorporated into the peptide. The selected class I and class II epitopes were joined by using the pertinent linkers. Several immuno-informatics tools were used to evaluate and validate the vaccine's capability in immunotherapy against ovarian cancer. The purpose of the study was to employ computational approaches along with reverse vaccinology to develop multi-epitopes subunit vaccination (MEV) for ovarian cancer treatment that can enhance cellular and humoral immunogenicity. Although the proposed multi-epitope vaccine model has the potential to impact the development of a prospective vaccine, further research is needed. They conclude that further laboratory testing and subsequent pharmacological trials are required to demonstrate its efficacy and potential. The complete workflow of the study is illustrated in Fig. 1.

Graphical representation of the methodology used in the current study (Color figure online)
Methodology
Sequence Retrieval and Preparation
Six cancer-testis antigens in ovarian cancer NY-ESO-1, AKAP4, ACRBP, PIWIL3, LAGE-1 were taken as targets. Their amino acid sequence was retrieved from Uniprot. Vaxijen v.2.0 online tool (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was utilized to identify the proteins having strong antigenicity. This tool evaluates the antigenic properties based on the alignment-free method and chiefly analyzes their physiochemical properties (Doytchinova and Flower DRJBb. 2007). Vaxijen v.2.0 requires protein sequence in Fasta format. Therefore, protein sequences in Fasta format were added, and the threshold was set at 0.5. Finally, proteins showing acceptable antigenicity and physiochemical properties were tested for similarity index with human proteins; for this purpose, Blastp was utilized.
T-cell Epitope Recognition and Evaluation
T-cells epitopes prediction is the significant step to design a vaccine, as it not only makes the whole procedure cost-effective but also, in comparison to the laboratory experiments, lessens the time required for the entire protocol (Saha 2006; Zhang et al. 2004). To identify the T-cell epitopes, the immune epitope database analysis resource (IEDB-AR) version 2.22 (https://www.iedb.org/) (Vita et al. 2018) was employed. This online server provides many tools for the estimation and assessment of antigenic epitopes. Protein sequences were submitted in Fasta format, consensus method was elected as prediction method, the human was chosen as source species, and all offered alleles were selected to forecast epitopes. Entire forecasted epitopes were additionally investigated from side to side diverse online bioinformatics tools, i.e., VaxiJen v2.0 was engaged to assess antigenic character, IEDB-AR v.2.22 MHC-I immunogenicity tool was employed for recognizing immunogenicity, AllergenFP v1.0 (Dimitrov et al. 2014) was utilized for inquiring allergenic character, and Toxin Pred online tool (Yadav et al. 2020) was betrothed to calculate the toxicity of the anticipated epitopes. AllergenFP server computes descriptor-based alignment-free fingerprint protocol to recognize the allergenic character of peptides with 88.9% accuracy (Dimitrov et al. 2014). Toxin Pred online tool involves machine-learning with a quantitative matrix to predict toxicity (Gupta et al. 2013). The transmembrane nature of epitopes was evaluated by the TMHMM server (Chen et al. 2003).
B-cell Epitope Recognition and Evaluation
Recognition of B-Cells Epitopes (BCEs) is a vital step in designing MEVs as it releases antibodies that lead to the generation of humoral immunity. Linear BCEs were identified via ABCPred online server (http://crdd.osdd.net/raghava/abcpred/). It employs a neural networking-based methodology to recognize BCEs (Saha 2006). The threshold and amino acid length were fixed to 0.5 and 14, respectively. Finally, the selected BCEs were evaluated for antigenic, allergic, immunogenic, and toxic profiles through vaxiJen v2.0, AllergenFP v1.0, IEDB-AR v.2.22 MHC-I immunogenicity, and ToxinPred, respectively.
Peptide Modeling and Molecular Docking Studies
To evaluate binding efficiency, the selected HTL and CTL epitopes were subjected to the PEP-FOLD v3.0 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) for the prediction of the peptide structure by utilizing the sOPEP sorting scheme with 200 simulations (Lamiable et al. 2016). HLAs crystal structures have been downloaded from RCSB PDB (https://www.rcsb.org/) (Berman et al. 2003). Through ClusPro (https://cluspro.bu.edu/login.php), docking was performed. To create and visualize the docked complex, the PyMOL molecular graphic system v.1.3 was used (Pymol 2002).
Estimation of Population Coverage
HLA allele distribution differs between various cultural groups and geographical constituencies of the Sphere, making population coverage an important parameter in constructing an MEV. The current study calculated collective population coverage of shortlisted HTL and CTL epitopes with identical HLA alleles with an IEDB population coverage tool (Vita et al. 2019).
Creation of Multi-Epitope Vaccine Sequence
Immuno-informatics tools were used for designing the vaccine sequence. To design the vaccine, the CTL and HTL high-scoring epitopes were linked with the correlated linkers. Furthermore, GM-CSF was supplemented as an adjuvant on the N-terminal to enhance its immunogenicity.
Host Homology Analysis
Homology to host protein can prompt autoimmunity. NCBI BLASTp online server (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) was utilized to determine the homology of constructed MEV with the human proteome (Altschul et al. 1990). The primary sequence of the vaccine’s protein was added in FASTA format, and Homo sapiens (taxid:9606) was taken as a target organism, and BLAST was run without changing any other parameter.
Evaluation of Allergenicity, Antigenicity and Physiochemical Properties
Different online tools were adopted to evaluate the final vaccine construct for physicochemical properties, antigenicity, and allergenicity. VaxiJen v2.0 was accessed to predict the antigenic behavior of the MEV construct, whereas AllerTop v2.0 (http://www.ddg-pharmfac.net/AllerTOP/) was adopted to evaluate its allergenicity. AllerTop includes the alignment-free estimation of the allergenic character of a protein by assessing its chief physicochemical properties (Dimitrov et al. 2017). Protparam tool of Expasy was used to recognize different physiochemical effects of the final MEV protein, for instance, theoretical pI, Grand Average of Hydropathicity (GRAVY), aliphatic index, stability index, molecular weight, and expected half-life for mammal cells, yeast, and E. coli. Solubility was predicted by the SolPro tool (Magnan et al. 2009).
Secondary Structure Prediction
SOPMA was used to predict the secondary structure of the vaccine’s protein (Deléage 2017). This tool measured the level of extended strands, alpha helices, random coils, and beta-turn. Two graphs were included in the results. One shows the prediction, and the alternative shows score curves for all expected positions.
Prediction of Tertiary Structure
For the prediction of the tertiary structure of the MEV, I-TASSER was used (Zhang YJBb. 2008). Using computer algorithms, the 3D model of protein can be generated from the amino acid sequence. Based on iterative template fragment assembly simulations and multiple-threading alignments, this server predicts three-dimensional protein structures. I-TASSER offers confidence scores for the determination of the accuracy of the model predicted.
Refinement and Validation of 3D Vaccine Construct
The GalaxyRefine web server (http:/galaxy.seoklab.org/) was used to modify the forecasted exemplary of the vaccine (Ko et al. 2012). The first, GalaxyRefine web-server reconstructs the side chain and then performs the structural repacking and the overall structural relaxation by using molecular dynamics. The ProSA-web, ERRAT server, and Ramachandran plot analysis were used to identify errors in the predicted tertiary structure (Wiederstein and Sippl MJJNar. 2007; Xia et al. 1992; Lovell et al. 2003). ProSA-web provides the typical quality score of the input construction. Moreover, for the evaluation of not-linked atom–atom interactions, the ERRAT server was used. Conclusively, the Ramachandran plot was used to evaluate energetically forbidden and permitted angles at dihedral angles psi and phi.
Estimation of B-Cells Epitopes
Both linear BCEs and conformational BCEs of constructed MEV were identified using the ABCPred online server and Ellipro tool of IEDB-AR v.2.22. In the ABCPred server, the MEV amino acid sequence was given as input, the threshold was set at 0.5, and the amino acid length was fixed at 14. In contrast, all parameters of the Ellipro tool were set by default, and the MEV 3D structure was given as input. PyMOL was employed to visualize the discontinuous epitopes in the final MEV construct (DeLano WLJCNopc. 2002).
Disulphide Engineering
Disulfide engineering is a new method of adding disulfide bonds between amino acids of a protein structure. A disulfide bond is a covalent bond that imparts significant firmness to a protein structure by authorizing accurate geometric conformations. Disulfide by Design v2.12 online tool was employed for disulfide engineering of MEV protein. Residue-pair search was done using a refined MEV structure, possible residue pairs were recognized for mutation, and cysteine residue was taken as a concluding target (Chen et al. 2003). Residues having an energy score lesser than 2.2 kcal/mol and χ3 angle within a range of − 87 and + 97° were considered for the formation of disulfide linkages (Craig and Dombkowski AAJBb. 2013).
Immune Simulation of Vaccine Construct
In-silico immune simulation was performed through C-ImmSim 10.1 server to validate the designed MEV's multi-epitope vaccine immunological responses. 3 constituents of the efficient mammal structures, such as the Thymus, lymph node, and bone marrow, were stimulated by C-ImmSim (Rapin et al. 2010). A minimum 30-day interval between two doses was considered. Three injections were administered in-silico with time stages of 1, 84, and 168, respectively, where one-time step equals 8 h in real life. The maximum value was set to 1000 simulation steps, while other stimulation parameters were maintained by default.
Codon Optimization and In Silico Cloning
As soon as the properties and in-silico immune simulations of fabricated MEV were examined prudently, Java Codon Adaptation Tool was used to optimize the codons (Grote et al. 2020). Codon optimization is a procedure commonly utilized to ameliorate the gene expression and enhance the translational efficiency of a gene of interest by inserting codon biased to the host organism (Burgess-Brown et al. 2008). Codon optimization of MEV was done in E. coli K12 prokaryotic expression system. Three available options were chosen to identify: (1) cleavage sites for restriction enzymes, (2) binding sites for prokaryote ribosome, and (3) rho-independent transcription termination. GC content and Codon adaptation index (CAI) values were noted and checked whether they fall under the acceptable range or not, which was 30–70% for GC content and < 0.8 for CAI (Sharp 1987). In addition to that, constraint and cloning of the adapted MEV sequence were accelerated by adding BamHI and HindIII restriction sites at its C and N-terminals correspondingly. This altered sequence was then taken for in silico cloning by means of the SnapGene v4.2 tool (https:/snapgene.com/). Cloning was completed in vector E. coli pET30a ( +) to authenticate the expression of sequence in-vitro systems.
Results
Sequence Retrieval and Preparation
Amino acid sequences of six cancer-testis antigens in ovarian cancer were retrieved from Uniprot. Proteins names and accession no were enlisted in Table 1. The antigenicity of all of these proteins was assessed by VaxiJen v.2.0, and the outcomes presented that all proteins were antigenic. Therefore, the sequence of these proteins was submitted to Blastp to identify their similarity with human proteins. The findings from Blastp showed that all proteins have no noticeable resemblance to human proteins; hence these proteins were chosen for further analysis.
T-Cell Epitope Recognition and Estimation
IEDB consensus method was adopted to forecast T-cell (including MHC-I and MHC-II) epitopes of target proteins. Owing to their strong defensive capabilities, epitopes showing interactions with more than one allele are taken as the most appropriate epitopes. The conservancy of predicted epitopes within the protein sequences was determined through the IEDB conservancy analysis tool. Their allergenicity and antigenicity were evaluated by Allergen FP 1.0 and Vaxijen. In total, 76 CTL epitopes (NY-7, AK-25, AC-10, PI-27, and LA-7) (Additional File 1: Table S1) and 37 HTL epitopes (NY-1, AK-11, AC-4, PI-19, and LA-2) were shortlisted (Additional File 1: Table S2).
B-Cell Epitope Recognition and Evaluation
Linear/continuous B cell epitopes (LBL) of all target proteins were identified by the ABCPred server. Just like T cell epitopes, selected LBL were antigenic, non-allergenic, and 100% conserved. Based on this criterion, 160 LBL epitopes (NY-11, AK-48, AC-36, PI-14, and LA-51) were predicted (Additional File 1: Table S3).
Screening of Epitopes
The criteria intended to include the epitopes in the MEV was that they should be 100% conserved among proteins, should be significantly antigenic/immunogenic, should be non-toxic and non-allergenic, should not be within the glycosylation sites and post-translation modification sites of the particular protein, and having no transmembrane helices. Therefore, only those epitopes satisfying the criteria mentioned above were selected for further analyses. Total 14 CTL epitopes (NY-3, AK-3, AC-3, PI-2, and LA-3) and 5 HTL epitopes (NY-1, AK-1, AC-1, and PI-2) were chosen for MEV designing (Table 1). We have selected that CTL and HTL epitopes for vaccine construction which overlap B cell epitopes.
Docking Between HLA Alleles and Epitopes
Structures of epitopes were predicted via the PEPFOLD server. Molecular docking was performed via the ClusPro server to analyze the interaction between the HLA alleles and selected cell epitopes. It has been observed that all the identified epitopes are attached deeply within the binding pockets of their respective alleles (Fig. 2). To be used in an MEV construct, all of the chosen epitopes displayed substantial binding ability and appropriateness to be used in the vaccine construct.

Docking of the selected epitope with Tcell receptors. Purple color showing receptor and yellow color showing the epitope
Population Coverage Analysis
The HLA alleles’ distribution differed among different countries and various geographical constituencies of the world. Population Coverage is a compulsory and vital constraint to identify vaccine validation. Herein, we calculated class I and class II epitopes’ population coverage along with their respective alleles. Global population coverage of the selected epitopes was found 97.59%. The highest population coverage was 98.62% observed in South Asia, followed by Europe and North America, where coverage was reported as 98.47% and 97.97%, respectively (Fig. 3).

Population coverage of the selected (Class I & Class II) epitopes along with their alleles and highly significant areas shown in this graph
Multi-Epitope Peptide Vaccine (MEV) Designing
The MEV construct was designed using all selected epitopes. Two motifs, including AAY and HEYGAEALERAG, were used to connect CTL epitopes. Moreover, via a GPGPG linker, the predicted HTL epitopes were combined. Adjuvants were attached using linkers. The final MEV construct was found to consist of 547 amino acids, including adjuvants and linkers (Fig. 4).

Schematic diagram of the constructed MEV structure having 547 amino acids, adjuvant at N-terminal, 14 (class I), and 5 (class II) epitopes
Immunogenic and Physiochemical Profiling
The construction of vector is based on the structure for both physical and immunol properties and characteristics. Homology was first checked against the human proteomics available data, and the outcomes thus obtained were contained to have no similarity with any region of human proteomics. Secondly, MEV was evaluated for antigenicity, toxicity, and allergenicity. This analysis found that constructed MEV is extremely antigenic with an antigenic score of 0.6299 (at threshold = 0.5), potentially harmless. Moreover, the physiochemical properties of MEV were also evaluated using Protparam online tool. Vaccine protein was found to be stable with a Molecular weight of 60,453.00 Da. Its theoretical pI and GRAVY were reported to be 6.09 and − 0.421 (negative sign shows hydrophilic nature), respectively. The mean half-life of MEV was measured, E. coli and yeast cell respectively. MEV was found to be soluble with a probability score of 0.864380. All these properties showed that the constructed MEV has an outstanding potential to act as a vaccine.
Prediction of Secondary Structure
The secondary structure of the MEV was studied with SOPMA. Predictions of SOPMA depend on vaccine sequence amino acids. Among 547 amino acids, 312 amino acids are involved in the α-helix formation, which represents 57.04%, 47 in β-strands formation, 8.59%, and coils are molded by 171 amino acids which represent 31.26% of the entire vaccine construct.
3D Structure Prediction and Refinement
To predict the tertiary structure of the designed MEV, I-TASSER was used, and the c-score was − 1.35. Model number 3 has been identified as the excellent-optimized model based on the different parameters used in the GalaxyRefine server that have RMSD, Clash score, Rama favored, etc., in comparison to the other models. This selected model has been chosen as the finalized vaccine model and the rough model that has undergone structural evaluation (Fig. 5A). The refined model's z-score has been calculated to be − 6.09 (Fig. 5C). According to the Ramachandran plot analysis, 85.1% residues were in the favored region, 3.0% in the disallowed region, 0.9% in the highly allowed region, and 11.0% in the allowed region (Fig. 5B). Moreover, the refined model ERRAT score was 82.4953.

A 3D structure of the vaccine construct showing α-helix (blue), β-strands (purple), and loops (yellow). B Ramachandran plot showing the 85.4% residues were in the favorable region. C This graph is showing the Z-Score of the vaccine construct (Color figure online)
Disulphide Engineering for Vaccine Stability
Disulfide by design v2.0 was used to carry out disulfide engineering to enhance the stability of MEV’s refined model. A total of 25 residue pairs could be used for this purpose. Nonetheless, four residue pairs showed energy and Chi3 value within the normal range. Therefore, it was finalized for disulfide engineering purposes. Consequently, eight mutations were introduced in the residue pair, i.e., Ala 171-Gly 205 with the energy of 1.36 kcal/mol and Chi3 value of + 73.04, Gly 205-Arg 210 with the energy of 0.58 kcal/mol and Chi3 value of + 90.90, Glu 209-Tyr 265 with the energy of 1.86 kcal/mol and Chi3 value of + 85.94, and Trp 253-Ser 261 with the energy of 1.81 kcal/mol and Chi3 value of − 68.32. (Fig. 6).

Protein stability is enhanced by Disulfide engineering. Total 4 amino acid residues were selected based on their energy, B-factor, and Chi3 value. Mutated residues are shown in grey and magenta colors (Color figure online)
Estimation of B Cell Epitopes
Besides releasing cytokines, B-lymphocytes also provide humoral immunity by producing antibodies. Hence, MEV should ideally have B cell epitopes with its domains. Thirty-eight linear B cell epitopes (Additional File 2: Table S1) and eight conformational B cell epitopes (Additional File 2: Table S2) were predicted from finalized MEV construct without altering ABCPred 2.0 and Ellipro prediction parameters. PyMOL facilitated visualizing the Conformational B cell epitopes in the 3D structure of the MEV construct (Fig. 7).

Identified conformational B-cell epitopes from MEV (Magenta) are shown in orange color (Color figure online)
Immune Simulation of Vaccine 3D Structure
A real immune system's immune responses result from a threshold response produced by all primary and secondary immunity responses. In silico host, immune system reaction to the antigen is shown in Fig. 8. IgG + IgG and IgM concentrations are continually elevated in the primary immune response preceded by IgG1 + IgG2, IgM, and IgG1 at the equally primary and secondary immune responses accompanying antigen reduction. Furthermore, to evaluate the vaccine, cytokine and interleukin responses were also observed. All the above analysis revealed that the vaccine has a decent immune response after subsequent encounters.

In-silico Immune response of the MEV as an antigen. A Antibodies, B Cytokines and interleukins
In-Silico Cloning
To guarantee the translation of MEV infrequently used E.coli hosts, in silico cloning was accomplished. The JCat server was used to refine MEV codons following the E.coli (strain K12) expression system before cloning. The optimized MEV sequence comprises 1641 nucleotides, a CAI value of 0.98, and a GC content of 56.00%, showing a high potential for reliability and positive protein expression. In-silico cloning was executed to validate the expression of MEV in a host cell. This stride was supported by the supplementation of two restriction sites (BamHI and HindIII) to ends of the MEV optimized nucleotide sequence. This modified sequence was then cloned to the pET30a ( +) vector at the multiple cloning sites (Fig. 9). The entire clone had a size of 7052 bp.

In-silico cloning was performed for optimized MEV into E. coli K12 expression system. In black color, the plasmid backbone is shown. The inserted DNA sequence is represented in purple color
Discussion
Ovarian cancer is still the deadliest of all gynecologic cancers. Lack of effective screening strategies results in 75 percent of women being diagnosed with ovarian cancer at an advanced stage with a survival rate of 46 percent five years after diagnosis (Lheureux et al. 2019). A typical treatment plan for ovarian cancer involves surgery and chemotherapy. Over 80 percent of patients will respond to initial therapy, but the majority will recur and require additional therapy. A major obstacle in the treatment of recurrent ovarian cancer is chemotherapy-resistant disease, which develops over the course of multiple therapies (Schaar et al. 2018). This demonstrates the need for new therapeutic interventions, including the development of immunotherapy for the ovarian cancer treatment. Cellular therapies, immune checkpoint inhibition, and cancer vaccines are some of the current immunotherapeutic options. Vaccine-based therapies aim to stimulate innate and adaptive immune responses against ovarian cancer-associated antigens.
Immune responses are crucial in the fight against malignancies and viral infections. An antigenic epitope is a fundamental unit that induces either a cellular or a humoral immune response. Therefore, a multi-epitope vaccine composed of a series of or overlapped peptides would be a good option for treating and preventing viral or tumors infections. Ideally, a multi-epitope vaccine would include epitopes that elicit B-cell, T-helper (Th), and CTL responses against a targeted virus or tumor (Buonaguro L, Consortium H 2016). The immune-informatics tools will provide valuable facilities for the accurate design of vaccines to achieve this aim. Different computational studies have demonstrated the viability, precision, and speed of in-silico tools for the study and design of multiple epitopes for various chronic disorders, including cancer (Pourseif et al. 2005, 2018).
The vaccine’s effectiveness is determined by the selection of correct CTL epitopes as the chief elements, adjuvant and HTL epitopes (Burg et al. 2006). Moreover, to produce protein having our desired functionality, we are required to fuse these components. We can also say that a protein may have various characteristics by choosing appropriate linkers. Therefore, suitable linkers must be utilized for the development of a peptide vaccine. In general, when administered with appropriate adjuvants, peptide vaccines in nature induce antigen-specific T-cell responses. More importantly, the adjuvant is considered a crucial constituent inside a peptide vaccine to maximize dendritic cells’ functioning and create an effective immune reaction.
The vaccine was designed by predicting epitopes from targeted cancer testes antigens i-e NY-ESO-1, AKAP4, ACRBP, SPAGE9, MAGEA9, CT45A3, PIWIL3, LAGE-1. Adjuvants must be added to multi-epitope vaccines to achieve sufficient immunogenicity (Meza et al. 2017). The ability of GM-CSF-based vaccines in preclinical studies for inducing a strong response against viruses and tumors has been demonstrated (Ahlers et al. 2002). This ability helps activate the dendritic cells by GM-CSF induced anti-tumor immunity mechanism and connects T cells with dendritic cells. Consequently, the anti-tumor response can be generated via GM-CSF effectively by using GM-CSF as an immune adjuvant in the candidate (Yan et al. 2017).
The structural and functional activities of a protein-based vaccine are mainly dependent on linkers. In the current study, different linkers were utilized for vaccine development. Two selected motifs, including AAY and HEYGAEALERAG, were used to connect CTL epitopes. The HEYGAALERAG sequence provides a particular site for the cleavage of systems responsible for proteasomal & lysosomal degradation. Besides, through the AAY sequence, the epitope presentation is improved. After cleavage, the C-terminal binds to chaperon transporters like TAP and others (Bergmann et al. 1996; Gu et al. 2013). Moreover, the T-helper components predicted in the study were joined together via a GPGPG linker. The GPGPG linkers induce conserve conformational dependent immunity and also stimulate antibodies HTL responses. The EAAAK is another sequence acting as linkers for monitoring molecular distances and for minimizing domain interference. Eventually, the current vaccine construct was built by linking adjuvant, HTL epitopes, and CTL epitopes as the main components of the vaccine (Livingston et al. 2002).
The molecular weight of the final constructed vaccine is 42.15 KDa, and it shows no homology with any portion of the human proteome. Furthermore, recombinant MEV protein showed excellent solubility upon overexpression in the E.coli host, making it easy access to the host (Khatoon and Pandey 2017). The instability index depicted that protein has a stable nature upon expression, augmenting the usage capacity further. The theoretical pI of 9.39 shows that the MEV is strongly alkaline and consistently connects in the physiological pH range. Moreover, the aliphatic index and GRAVY score represented the thermo-stable and hydrophobic nature, respectively (Ismail et al. 2020; Ikram et al. 2018; Tahir ul Qamar M, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F,, et al. 2020).
The HLA alleles are so significant since they hold the responses to T-cell epitopes, nevertheless, grounded on background, these alleles astonishingly polymorphic. The T-cell epitopes are expected to depict binding affinity with many HLA alleles so that more population coverage can be achieved. The outcomes demonstrated that the selected epitopes and their respective alleles are found in major geographical areas of the World with global population coverage of 97.59%.
The modeling of 3D structure provides outstanding support in the estimation of protein dynamics, function, and interaction capabilities with other proteins. Therefore, the 3D structure of MEV was predicted, and its desirable properties were considerably increased through refinement. Ramachandran plot analysis, ERRAT quality factor, and z-score verified the excellent quality of the model. B-cell epitopes were predicted from the MEV construct to determine whether it contained enough epitopes for antibodies to detect and latch onto (Flaviviruses 1990).
The immunoreactivity testing through serological assessment is a fundamental step to authenticate a vaccine construct (Gori et al. 2013). Expression of the foreign genes may differ inside the genome of a host cell. This variation is the inconsistency of the mRNA codon; therefore, it is required to optimize the codon to ensure a higher expression level in the host cell (Pandey et al. 2018). CAI (58.46) and GC (1.0) content of optimized codon showed promising outcomes for higher protein expression in E. coli expression system, which are extensively used for the synthesis of recombinant protein (Chen RJBa. 2012; Rosano 2014).
In the recent study, the MEV construct was designed using the next-generation vaccine approach, which is skilled in producing a threshold immunity reaction against ovarian cancer. Furthermore, the active immune response has been observed in real life in immune stimulation. Further, wet lab inquiries are highly needed to validate its actual potential to combat ovarian cancer.
Conclusion
The proposed multi-epitope vaccine model, when combined with immunological data and computational analysis, may have an impact on the development of a prospective vaccine. We conclude that an estimated amount of vaccine model will pose a positive effect on treating ovarian cancer. Moreover, the present work is an outcome of an integrated vaccinomics approach. Thus, laboratory testing and subsequent pharmacological trials are required to prove the efficacy and potential of the developed MEV.
Data Availability
All data and materials generated or analyzed during this study were included in the published article.
Abbreviations
- MHC:
-
Major histocompatibility complex
- MEV:
-
Multiepitope-based subunit vaccine
- GM-CSF:
-
Granulocyte–macrophage-colony-stimulating factor
References
Aguilera R, Saffie C, Tittarelli A, González FE, Ramírez M, Reyes D et al (2011) Heat-shock induction of tumor-derived danger signals mediates rapid monocyte differentiation into clinically effective dendritic cells. Clin Cancer Res 17:2474–2483
Ahlers JD, Belyakov IM, Terabe M, Koka R, Donaldson DD, Thomas EK et al (2002) A push–pull approach to maximize vaccine efficacy: abrogating suppression with an IL-13 inhibitor while augmenting help with granulocyte/macrophage colony-stimulating factor and CD40L. Proc Natl Acad Sci 99:13020–13025
Altschul SF, Gish W, Miller W, Myers EW (1990) Lipman DJJJomb. Basic Local Alignment Search Tool 215:403–410
Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM (1984) Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation
Bergmann CC, Yao Q, Ho C-K (1996) Buckwold SLJTJoI. Flanking Residues Alter Antigenicity and Immunogenicity of Multi-Unit CTL Epitopes 157:3242–3249
Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide protein data bank. Nat Struct Mol Biol 10:980
Buonaguro L, Consortium H (2016) Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol Immunother 65:93–99
Burgess-Brown NA, Sharma S, Sobott F, Loenarz C et al (2008) Codon Optimization Can Improve Expression of Human Genes in Escherichia Coli: A Multi-Gene Study 59:94–102
Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A et al (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 9:e1003266
Chen Y, Yu P, Luo J, Jiang Y (2003) Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome 14:859–865
Chen RJB (2012) Bacterial expression systems for recombinant protein production: E coli and beyond. Biotechnol Adv 30:1102–1107
Coburn S, Bray F, Sherman ME (2017) Trabert BJIjoc International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Inter J Cancer 140(2451):2460
Craig DB, Dombkowski AAJBB (2013) Disulfide by Design 20: a web-based tool for disulfide engineering in proteins. BMC Bioinformat 14:1–7
Danilova A, Misyurin V, Novik A, Girdyuk D, Avdonkina N, Nekhaeva T et al (2020) Cancer/testis Antigens Expression during Cultivation of Melanoma and Soft Tissue Sarcoma Cells 10:3
DeLano WLJCNopc (2002) Pymol: An open-source molecular graphics tool. 40:82–92.
Deléage GJB (2017) ALIGNSEC: viewing protein secondary structure predictions within large multiple sequence alignments
Dimitrov I, Naneva L, Doytchinova I, Bangov IJB (2014) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30:846–851
Dimitrov I, Bangov I, Flower DR, Doytchinova IJJomm, (2014) AllerTOP v 2—a server for in silico prediction of allergens. J Mol Modeling 20:1–6
Doytchinova IA, Flower DRJBB (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:1–7
Edwards HM, Noer MC, Sperling CD, Nguyen-Nielsen M, Lundvall L, Christensen IJ et al (2016) Survival of ovarian cancer patients in Denmark: Results from the Danish gynaecological cancer group (DGCG) database 1995–2012. Acta Oncol 55:36–43
Faramarzi S, Ghafouri-Fard SJI (2017) Melanoma: a prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 9:1103–1113
Monath TP. Flaviviruses (1990) Army Medical Research Inst Of Infectious Diseases Fort Detrick MD
Garcia-Soto A, Lucci J, Podack E, Schreiber T, Schroeder EJGO (2014) Cancer-Testis Antigen Expression is Shared between Epithelial Ovarian Cancer Tumors 133:95
Garcia-Soto AE, Schreiber T, Strbo N, Ganjei-Azar P, Miao F, Koru-Sengul T et al (2017) Cancer-Testis Antigen Expression is Shared between Epithelial Ovarian Cancer Tumors 145:413–419
Gjerstorff MF, Andersen MH, Ditzel HJJO (2015) Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget 6:15772
Gori A, Longhi R, Peri C, Colombo GJAA (2013) Peptides for Immunological Purposes: Design, Strategies and Applications 45:257–268
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC et al (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526–W531
Gu SH, Nicolas V, Lalis A, Sathirapongsasuti N, Yanagihara RJI, Genetics E (2013) Complete genome sequence and molecular phylogeny of a newfound hantavirus harbored by the Doucet’s musk shrew (Crocidura douceti) in Guinea. Infect Genet Evol 20:118–123
Guleria S, Jensen A, Toender A (2020) Kjaer SKJCC. Control Risk of Epithelial Ovarian Cancer among Women with Benign Ovarian Tumors: a Follow-up Study 31:25–31
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP et al (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8:e73957
Hajighahramani N, Nezafat N, Eslami M, Negahdaripour M, Rahmatabadi SS, Ghasemi Y (2017) Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect Genet Evol 48:83–94
Henriksen JR, Donskov F, Waldstrøm M, Jakobsen A, Hjortkjaer M, Petersen CB et al (2020) Favorable Prognostic Impact of Natural Killer Cells and T Cells in High-Grade Serous Ovarian Carcinoma 59:652–659
Ikram A, Zaheer T, Awan FM, Obaid A, Naz A, Hanif R et al (2018) Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci Rep 8:1–14
Ismail S, Ahmad S, Azam SSJJoML, (2020) Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J Mol Liquids 314:113612
Jordan SJ, Green AC, Whiteman DC, Webb PM, Study AC (2007) Risk factors for benign, borderline and invasive mucinous ovarian tumors: epidemiological evidence of a neoplastic continuum? Gynecol Oncol 107:223–230
Kar PP, Srivastava A (2018) Immuno-informatics analysis to identify novel vaccine candidates and design of a multi-epitope based vaccine candidate against Theileria parasites. Front Immunol 9:2213
Khalid H, Ashfaq UA (2020) Exploring HCV genome to construct multi-epitope based subunit vaccine to battle HCV infectio: Immunoinformatics based approach. J Biomed Informat 108:1034
Khatoon N, Pandey RK (2017) Prajapati VKJSr. Exploring Leishmania Secretory Proteins to Design B and T Cell Multi-Epitope Subunit Vaccine Using Immunoinformatics Approach 7:1–12
Ko J, Park H, Heo L (2012) Seok CJNar. GalaxyWEB Server for Protein Structure Prediction and Refinement 40:W294–W297
Kulkarni P, Uversky VNJIjoms, (2017) Cancer/testis antigens:“smart” biomarkers for diagnosis and prognosis of prostate and other cancers. Int J Mol Sci 18:740
Kurman RJ (2016) Shih I-MJTAjop. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded 186:733–747
Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P (2016) PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44:W449–W454
Lheureux S, Gourley C, Vergote I, Oza AM (2019) Epithelial ovarian cancer. The Lancet 393:1240–1253
Li Y, Li J, Wang Y, Zhang Y, Chu J, Sun C et al (2017) Roles of cancer/testis antigens (CTAs) in breast cancer. Cancer Lett 399:64–73
Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J et al (2002) A Rational Strategy to Design Multiepitope Immunogens Based on Multiple Th Lymphocyte Epitopes 168:5499–5506
Lovell SC, Davis IW, Arendall WB III, DeBakker PI, Word JM, Prisant MG et al (2003) Structure Validation by Cα Geometry: φ, ψ and Cβ Deviation 50:437–450
Magnan CN, Randall A, Baldi P (2009) SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 25:2200–2207
Mahmood M, Javaid A, Shahid F, Ashfaq UA (2021) Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect Genet Evol 91:104795
Mamede LD, de Paula KG, de Oliveira B, Dos Santos JSC, Cunha LM, Junior MC et al (2020) Reverse and structural vaccinology approach to design a highly immunogenic multi-epitope subunit vaccine against Streptococcus pneumoniae infection. Infect Genet Evol 85:1044
Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S et al (2014) Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 11:509
Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C (2017) A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol 49:309–317
MT ul Qamar, Rehman A, Ashfaq UA, Awan MQ, Fatima I, Shahid F, et al. (2020) Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. BioRxiv
Qamar MT, Shahid F, Aslam S, Ashfaq UA, Aslam S, Fatima I et al (2020) Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect Dis Poverty 9:1–14
Nezafat N, Eslami M, Negahdaripour M, Rahbar MR, Ghasemi Y (2017) Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol BioSyst 13:699–713
Pandey RK, Ojha R, Aathmanathan VS, Krishnan M, Prajapati VKJV (2018) Immunoinformatics Approaches to Design a Novel Multi-Epitope Subunit Vaccine against HIV Infection 36:2262–2272
Pourseif MM, Moghaddam G, Daghighkia H, Nematollahi A, Omidi YJBB (2018) A Novel B-and Helper T-Cell Epitopes-Based Prophylactic Vaccine against Echinococcus Granulosus 8:39
Pourseif MM, Moghaddam G, Naghili B, Saeedi N, Parvizpour S, Nematollahi A et al (2018) A novel in silico minigene vaccine based on CD4+ T-helper and B-cell epitopes of EG95 isolates for vaccination against cystic echinococcosis. Comput Biol Chem 72:150–163
DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography 2002;40:82–92.
Rapin N, Lund O, Bernaschi M, Castiglione FJPO (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5:e9862
Rosano GL (2014) Ceccarelli EAJFim. Recombinant Protein Expression in Escherichia Coli: Advances and Challenges 5:172
Saha S (2006) Raghava GPSJPS, Function, Bioinformatics. Prediction of Continuous B-Cell Epitopes in an Antigen Using Recurrent Neural Network 65:40–48
Schaar B, Krishnan V, Tallapragada S, Dorigo O (2018) Cell-based immunotherapy in gynecologic malignancies. Curr Opin Obstet Gynecol 30:23–30
Seledtsov V, Goncharov A (2015) Seledtsova GJHv, immunotherapeutics. Clinically Feasible Approaches to Potentiating Cancer Cell-Based Immunotherapies 11:851–869
Shahid F, Ashfaq UA, Javaid A, Khalid H (2020) Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect Genet Evol 80:1041
Sharp PM (1987) Li W-HJNar. The Codon Adaptation Index-a Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications 15:1281–1295
Siegel RL, Miller KD (2016) Jemal AJCacjfc. Cancer Statistics 2016(66):7–30
Tahir ul Qamar M, Shokat Z, Muneer I, Ashfaq UA, Javed H, Anwar F, et al (2020) Multiepitope-Based Subunit Vaccine Design and Evaluation against Respiratory Syncytial Virus Using Reverse Vaccinology Approach. Vaccines 8:288
Tio D, Kasiem FR, Willemsen M, van Doorn R, van der Werf N, Hoekzema R et al (2019) Expression of Cancer/testis Antigens in Cutaneous Melanoma: a Systematic Review 29:349–357
Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD et al (2018) Ovarian cancer statistics. CA: A Cancer J Clinicians 2018(68):284–296
van der Burg SH, Bijker MS, Welters MJ, Offringa R (2006) Melief CJJAddr. Improved Peptide Vaccine Strategies, Creating Synthetic Artificial Infections to Maximize Immune Efficacy 58:916–930
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR et al (2019) The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 47:D339–D343
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR et al (2018) The immune epitope database (IEDB). Nucleic Acids Res 47:D339–D343
Wiederstein M, Sippl MJJNar. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. 2007;35:W407-W10.
Xia TH, Bushweller JH, Sodano P, Billeter M, Björnberg O, Holmgren A et al (1992) NMR structure of oxidized Escherichia coli glutaredoxin: comparison with reduced E coli glutaredoxin and functionally related proteins. Protein Sci 1:310–321
Yadav S, Prakash J, Shukla H, Das KC, Tripathi T, Dubey VK (2020) Design of a multi-epitope subunit vaccine for immune-protection against Leishmania parasite. Pathogens and Global Health 114:471–481
Yan W-L, Shen K-Y, Tien C-Y, Chen Y-A (2017) Liu S-JJI. Recent Progress in GM-CSF-Based Cancer Immunotherapy 9:347–360
Yousefi H, Yuan J, Keshavarz-Fathi M, Murphy JF (2017) Rezaei NJEroci. Immunotherapy of Cancers Comes of Age 13:1001–1015
Zhang M, Ishii K, Hisaeda H, Murata S, Chiba T, Tanaka K et al (2004) Ubiquitin-fusion degradation pathway plays an indispensable role in naked dna vaccination with a chimeric gene encoding a syngeneic cytotoxic t lymphocyte epitope of melanocyte and green fluorescent protein. Immunology 112:567–574
Zhang YJBB (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinfo 9:1–8
Acknowledgements
We acknowledge Department of Bioinformatics and Biotechnology, Government College University Faisalabad for providing computational facilities to conduct this piece of work.
Funding
The study was conducted using resources available online and no additional funding was involved.
Author information
Authors and Affiliations
Contributions
UAA generated the idea of the detailed research work, designed and performed immuno-informatics work and review the draft. MS performed the immunoinformatics work, FS performed docking, FI revised the draft. AJ performed and described the immune simulation of vaccine construct.
Corresponding author
Ethics declarations
Conflict of interest
There is no financial and other conflict of interest related to this article.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10989_2021_10294_MOESM1_ESM.docx
Supplementary file1 Additional-file 1: Table S1: MHC Class I selected epitopes predicting through IEDB server using. Table S2: MHC Class II selected epitopes predicted through IEDB server using consensus method. Table S3: Predicted Linear B-Cells using ABCpred server. (DOCX 39 kb)
10989_2021_10294_MOESM2_ESM.docx
Supplementary file2 Additional file 2: Table S1: Selected linear B-cells of the MEV predicted by the Abcpred server. Table S2: Ellipro server is used to predict the conformational B-cells (DOCX 23 kb)
Rights and permissions
About this article

Cite this article
Sufyan, M., Shahid, F., Irshad, F. et al. Implementation of Vaccinomics and In-Silico Approaches to Construct Multimeric Based Vaccine Against Ovarian Cancer. Int J Pept Res Ther 27, 2845–2859 (2021). https://doi.org/10.1007/s10989-021-10294-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-021-10294-w