
Abstract
Matrix metalloproteinases (MMPs) degrade and modify the extracellular matrix (ECM) as well as cell-ECM and cell-cell contacts, facilitating detachment of epithelial cells from the surrounding tissue. MMPs play key functions in embryonic development and mammary gland branching morphogenesis, but they are also upregulated in breast cancer, where they stimulate tumorigenesis, cancer cell invasion and metastasis. MMPs have been investigated as potential targets for cancer therapy, but clinical trials using broad-spectrum MMP inhibitors yielded disappointing results, due in part to lack of specificity toward individual MMPs and specific stages of tumor development. Epithelial-mesenchymal transition (EMT) is a developmental process in which epithelial cells take on the characteristics of invasive mesenchymal cells, and activation of EMT has been implicated in tumor progression. Recent findings have implicated MMPs as promoters and mediators of developmental and pathogenic EMT processes in the breast. In this review, we will summarize recent studies showing how MMPs activate EMT in mammary gland development and in breast cancer, and how MMPs mediate breast cancer cell motility, invasion, and EMT-driven breast cancer progression. We also suggest approaches to inhibit these MMP-mediated malignant processes for therapeutic benefit.
Similar content being viewed by others


Matrix Metalloproteinases: Overview
There are 23 human MMPs (Degradome database; http://degradome.uniovi.es) [1], including 17 soluble, secreted enzymes and 6 membrane-associated enzymes (Fig. 1); they differ from each other in their structural domain architecture, in their substrate specificity, and in their temporal and tissue specific expression patterns. MMPs were originally named for their preferred substrates within the extracellular matrix (ECM): collagen-cleaving MMPs (MMP-1, -8, and -13) were designated collagenases, gelatin (denatured collagen)-cleaving MMPs (MMP-2 and -9) were termed gelatinases, and MMPs that degraded a broad spectrum of ECM proteins were called stromelysins (MMP-3, -10, and -11) or matrilysins (MMP-7). As the MMP family grew with the discovery of additional paralogs, including the membrane-associated MMPs, of which MT1-MMP/MMP-14 is the founding member, a numbering system was adopted, and MMPs are now grouped according to their domain structure.

MMP domain structure and protein fold. a The various domain organizations of human MMPs are illustrated; S, signal peptide; Pro, propeptide; CAT, catalytic domain; F, fibronectin repeats; PEX, hemopexin domain; TM, transmembrane domain; GPI, glycophosphatidylinositol membrane anchor; C, cytoplasmic domain; CA, cysteine array; Ig, immunoglobulin-like domain. The flexible, variable length linker or hinge region is depicted as a wavy black ribbon. b The protein structure of the domains of a representative proMMP (proMMP-2) is shown; the individual domains, colored as in the cartoon above, have been separated for visual clarity. Dotted lines indicate the coordination of prodomain cysteine to the catalytic zinc (gray sphere), as well as the points of covalent attachment between the catalytic domain and the prodomain, fibronectin repeats, and PEX domain. Figure was generated with Pymol [155], using coordinates from Protein Databank entry 1GXD [156].
MMPs are modular enzymes (Fig. 1a). The core functional domain of every MMP is the catalytic domain, a compact globular domain of 160–170 amino acids featuring a highly conserved HExxHxxGxxH zinc binding motif, responsible for chelating the catalytically essential zinc ion at the enzyme active site [2]. The catalytic zinc and substrate binding cleft of the catalytic domain comprise the MMP region targeted for binding and inhibition by the endogenous tissue inhibitors of metalloproteinases (TIMPs) [3], and also by the majority of small-molecule, synthetic pharmaceutical inhibitors of MMPs [4]. MMPs are produced as latent proenzymes, in which an N-terminal prodomain of ∼80 amino acids blocks catalytic activity by physically blocking the active site, through coordination of a conserved cysteine residue within a PRCGxPD motif (“the cysteine switch”) to the catalytic zinc [2, 5]. Upon stepwise interaction with and cleavage by one or more activating proteases [5, 6], the cleaved MMP prodomain dissociates from the catalytic domain, releasing the active enzyme.
Most MMPs also possess additional accessory domains that act to modulate catalytic activity, substrate recognition, and cellular localization [3, 7]; some accessory domains may also confer non-catalytic functions of potential significance for understanding MMP roles in EMT and tumor progression. The gelatinases MMP-2 and -9 are assisted in substrate binding of gelatin, collagens, and laminin by three fibronectin repeats inserted into the catalytic domain [8]. All human MMPs with the exception of MMP-7, -23, and -26A possess a C-terminal hemopexin (PEX) domain, a four-bladed propeller structure that is connected to the catalytic domain by a flexible linker [9, 10]. PEX domains have been shown to mediate the binding and unwinding of collagen triple helices by collagenases, facilitating cleavage by the MMP catalytic domain [11–14], as well as the recognition of other substrates including gelatin binding by MMP-9 [15], fibrinogen binding by MMP-2 [16], and targeting of several chemokines by MMP-2 [17–19]. Beyond substrate recognition, PEX domains can mediate interactions with tissue inhibitors of metalloproteinases (TIMPs), with distinctly different results for different MMP/TIMP pairs: proMMP-2 is targeted for MT1-MMP-mediated activation by TIMP-2 [3, 5], while proMMP-9, by binding to TIMP-1, is protected from activation [20–22].
In mammary epithelial cells, specific interactions between PEX domains and integrins or other cell surface receptors have been found to facilitate MMP activation, to localize soluble MMPs to sites of pericellular proteolysis, or to regulate MMP endocytosis and turnover [10]. MMP-9 docking to the hyaluronan receptor CD44 mediates proteolytic activation of TGF-β and promotion of tumor invasion and angiogenesis in a murine mammary carcinoma model [23]. Association of the MT1-MMP PEX domain with CD44H leads to localization of MT1-MMP at the leading edge of migrating cells [24] and facilitates cell migration [25]. Interaction between MT1-MMP and CD44 also stimulated epithelial cell self-sorting in an engineered model of mammary ductal morphogenesis; this function did not appear to depend upon MMP catalytic activity [26].
MMPs Stimulate Breast Cancer Progression
While a finely tuned array of MMPs is instrumental in orchestrating tissue development and homeostasis, the misregulation of MMPs is widespread in many pathological settings and especially in cancer, where MMP overexpression contributes to tumorigenesis and tumor progression through multiple mechanisms. MMP proteolysis serves a path-clearing role in facilitating the movement of cells or groups of cells through ECM [27, 28]; in this process, cleavage of some ECM components unmasks cryptic sites, generating fragments with new biological activities modulating migration, growth, or angiogenesis [27, 28]. MMPs also cleave cell-ECM adhesion proteins and cell-cell junction proteins, releasing individual epithelial cells from epithelial sheets, initiating outside-in signaling pathways that lead to widespread changes in gene transcription patterns, or generating soluble ectodomain fragments with novel activities. MMP-1 cleaves and activates the protease activated receptor-1 (PAR-1), leading to increased migration and invasion of breast cancer cells [29]. Targeting of E-cadherin by MMP-3 or MMP-7 generates a bioactive fragment that promotes invasion [30], and contributes to a cascade of molecular alterations leading to EMT in mammary epithelial cells [30, 31]. MT1-MMP processing of αv integrin enhances breast cancer cell migration [32, 33], and MMP shedding of the ectodomain of P-cadherin facilitates breast cancer cell invasion [34]. MMPs can also promote breast tumor progression by targeting soluble molecules. Examples include protease activation cascades (activation of MMP-9 by MMP-3 [22], activation of MMP-2 by MT1-MMP [35]), the activation of latent TGF-β by MMP-2 and MMP-9 [23], and the N-terminal truncation of interleukin-8 (IL-8) by MMP-9, increasing its neutrophil-activating potential by an order of magnitude [36].
Transgenic or knockout mouse models have been used to establish specific effects of individual MMPs on mammary tumor development (reviewed in [37]). MMP-3 overexpression driven by the whey acidic protein (WAP) promoter, most active in mammary gland epithelial cells from mid-pregnancy and during lactation, led to widespread premalignant alterations and spontaneous tumor formation [38]. Similarly, overexpression of MMP-7 under control of the mouse mammary tumor virus (MMTV) promoter, which is active during puberty and greatly enhanced during pregnancy, resulted in spontaneous formation of premalignant mammary hyperplasias, and accelerated tumor formation in bitransgenic MMTV-MMP-7/neu mice [39]. MMTV-driven overexpression of MT1-MMP also led to premalignant mammary gland abnormalities and spontaneous adenocarcinoma [40]. In studies with MMP-11 knockout mice subjected to 7,12-dimethylbenzanthracene (DMBA)-induced carcinogenesis, MMP-11 null mice developed fewer tumors [41]; MMP-11 null mice also developed fewer spontaneous tumors in the mammary gland cancer prone MMTV-ras model [42]. One clear conclusion from these studies is that some MMPs can act as tumor promoters in mammary carcinogenesis, impacting neoplastic risk from the very earliest stages of premalignant change [38, 43].
MMPs can also modulate later stages of cancer progression in genetic models of breast cancer. For example, knockout of MMP-9 in the MMTV-polyoma virus middle T-antigen (PyMT or PyVT) multistage mammary tumorigenesis model resulted in an 80% reduction in lung metastatic burden, indicating the importance of MMP-9 in metastasis and angiogenesis in this model [44]. In another study employing the MMTV-PyMT tumorigenesis model, MT1-MMP null mammary glands transplanted into syngeneic mice developed tumors with a markedly reduced capacity to metastasize to the lungs, compared with an MT1-MMP sufficient control group, demonstrating a role for tumor MT1-MMP in the metastatic process [45]. In a study investigating the roles of MMPs in recruitment of stromal bone-resorbing osteoclasts to breast-to-bone metastases, mammary tumor cells implanted into the bones of MMP-7 null mice formed smaller, slower growing tumors with recruitment of fewer osteoclasts and less osteolysis, implicating MMP-7 in this aspect of metastatic progression [46].
Of course, MMPs do not act as universal tumor promoters under all circumstances, and the effects observed can also vary depending upon the model and upon the genetic background of the mice [43, 44]. In contrast with the protumorigenic effects of MMP-3 in the WAP promoter model [38], MMTV-driven MMP-3 expression in a different strain of mice did not lead to spontaneous tumorigenesis, and in a DMBA chemical carcinogenesis protocol, MMTV-MMP-e mice were reported to have fewer mammary gland tumors and more apoptotic cells [43, 47]. In contrast with the tumor promoting effect of MT1-MMP in mice with mammary-directed overexpression of this protease [40], MT1-MMP had a growth suppressing effect in the MMTV-PyMT genetically induced model when tumorigenesis in MT1-MMP null versus MT1-MMP sufficient mammary glands were compared [45]. For MMP-9 promotion of breast-to-lung metastasis as well, the genetic background of the mice was a determining factor, as C57BL/6 mice showed MMP-9 dependent promotion of metastatic growth, whereas no significant differences were observed between wt and MMP-9 null mice of the FVB/N background [44]. These observations underscore the complexity of the process of tumor development, in which MMPs must interact with many other variables; it has been suggested that as in mice, genetic modifiers present in human patient populations may distinguish subgroups likely to benefit from therapeutic intervention in MMP-mediated processes [44].
MMPs and Physiological EMT
EMT is a process integral to the formation of many tissues and organs during development [48–51]. Activation of developmental EMT has been found to follow a defined sequence of events: morphogenesis of the epithelial tissue and specification of the cells that will undergo EMT, disruption or degradation of the basement membrane, breakdown of the epithelial tissue structure followed by ingression of the separated cells, and differentiation to the motile mesenchymal phenotype [49]. While MMPs have long been suspected to play roles in many different EMT-related tissue morphogenesis and cell migration processes, direct evidence of MMP involvement has been best characterized for neural crest delamination, endocardial cushion invasion, and mammary gland branching morphogenesis. EMT of the neural crest during embryogenesis releases mesenchymal cells that migrate through the body, giving rise to a wide variety of tissue types, including glial and neuronal cells, adrenal glandular tissues, melanocytes, and skeletal and connective tissues [49, 52] (Fig. 2a). MMP-2 becomes activated in the neural cells as they are undergoing EMT, but inactivated as the cells begin to disperse [53–56]; blocking MMP-2 inhibits EMT without affecting the migration of the detached neural crest cells [53]. EMT of embryonic endocardial cells into the endocardial cushion creates precursors of the valvular and septal structures (Fig. 2b) [57], and also is dependent upon expression of MMP-2, as treatment with MMP inhibitors blocks mesenchyme formation [58, 59]. Studies using endocardial cushion explants grown on collagen gels revealed that MMP-2-dependent EMT involves degradation of collagen-IV [58], and requires specific association of MMP-2 with integrin αvβ3 [60].

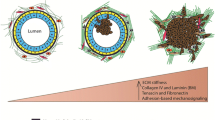
MMP-induced EMT in development. a. Neural crest delamination is facilitated by expression of MMP-2 in the cells undergoing EMT. b. EMT of endocardial cushions during early stages of heart development depends upon expression of MMP-2 in the endocardial cells. c. Secondary (2°) branching of the mammary ductal tree involves breakdown of epithelial structure, acquisition of invasive characteristics, and degradation of the basement membrane, processes shown to be dependent upon MMP-3, which is produced locally in response to morphogenic signals by the surrounding stromal cells.
Unlike many other tissues, the majority of mammary gland development occurs postnatally. During puberty, the rudimentary mammary gland grows into the fat pad through ductal extension and branching morphogenesis [61–63]. Extension of the ducts into the fat pad occurs at the endbuds, invasive structures that express high levels of EMT-associated transcription factors, including Snail and Twist [64] as well as MMP-2 and MT1-MMP [65]. Mammary branching morphogenesis occurs by two distinct mechanisms: primary branching through endbud bifurcation, and secondary branching, a process strikingly similar to developmental EMT, in which differentiated, ductal epithelium dedifferentiates, detaches from the adjacent epithelial cells, penetrates the basement membrane, and invades into the surrounding tissue (Fig. 2c). MMP-3 is a key mediator of secondary branch formation, as transgenic mice lacking MMP-3 expression have significantly reduced secondary branching, while the WAP-MMP-3 mice have increased secondary branching and ductal complexity [65, 66]. Upregulation of MMP-3 has also been implicated in the increased side-branching observed in transgenic mice in which retinoic acid signaling pathways are inhibited [67]. The mechanism by which MMP-3 induces branching morphogenesis has been investigated in 3D culture models in which mouse mammary epithelial cell clusters are grown in collagen I gels. These studies have shown that epimorphin, a stromal cell-produced morphogen, induces expression of MMP-3, and that this is both necessary and sufficient for activation of the branching process [68–71]. Activation of the fibroblast growth receptor signaling pathway, which has also been implicated in mammary branching morphogenesis, also induced MMP-3 expression and branch initiation in mammary epithelial cells grown in 3D collagen [72].
MMPs and EMT in Breast Cancer
MMPs have been associated with EMT in cancer progression through three distinct mechanisms: (a) elevated levels of MMPs in the tumor microenvironment can directly induce EMT in epithelial cells, (b) cancer cells that undergo EMT can produce more MMPs, facilitating cell invasion and metastasis, and (c) EMT can generate activated stromal-like cells that drive cancer progression via further MMP production. The most dramatic of these is MMP-dependent activation of the EMT program (Fig. 3a), seen in a variety of epithelial cell types, including kidney [73–76], ovary [77], lens [78], lung [79], and prostate [80], although MMP-induced EMT has been best characterized in mammary epithelial cells. Tumors that developed in the WAP-MMP-3 mice showed mesenchymal characteristics [38, 81, 82], and dissection of this process revealed that exposure of cultured mouse mammary epithelial cells to MMP-3 directly activates EMT [31, 83]. MMP-3 mediates these effects by stimulating increased expression of Rac1b [84], a constitutively activated splice variant of Rac1 found in breast and colorectal cancer cells [85–89], which in turn triggers EMT by increasing levels of cellular reactive oxygen species [84, 90]. While the process by which MMP-3 initiates these effects has not been completely defined, MMP-3 has been shown to cleave E-cadherin, promoting dissolution of epithelial cells and releasing a bioactive fragment of E-cadherin that induces cell motility [30, 31]. It is likely that many studies in which MMPs have been seen to stimulate cancer cell motility and invasion, although not directly investigating these phenomena in the context of EMT, have in fact been observing the cellular consequences of an incomplete activation of the EMT program. Unlike developmental EMT, where MMPs are a component of an organized morphogenic program, the chaotic and MMP-enriched tumor microenvironment induces an uncoordinated and incomplete EMT. As a consequence, these EMT-activated cells may acquire significant tumor-promoting abilities even as they retain many of their original characteristics, making it difficult to distinguish them from the original tumor mass from which they are derived.

MMPs facilitate EMT-associated tumor progression. a. Exposure of epithelial cells to MMPs can directly induce EMT. b. Increased expression of MMPs in cells which have undergone EMT facilitates cancer cell invasion. c. EMT can produce nonmalignant stromal cells which drive tumor initiation and progression through production of MMPs.
Breast cancer cells which have undergone EMT also show increased expression of MMPs, facilitating their invasive, metastatic characteristics (Fig. 3b; as this topic was comprehensively reviewed in 2005 [91], only highlights and more recent studies will be covered here). Early investigations revealed that breast cancer cell lines expressing mesenchymal markers often expressed MMPs, and that suppression of these MMPs blocked their invasive and migratory characteristics [92–94]. Subsequent investigations have identified MMP upregulation associated with a variety of EMT processes, although the specific MMPs induced seem to depend upon the nature of the EMT-inducing agent and the model system used. Transcriptional profiling studies of Ras-transformed mouse mammary epithelial cells induced to undergo EMT by treatment with TGFβ revealed MMP-2, MMP-12, and MMP-13 among the most upregulated transcripts [95, 96]. Culture of MCF10A cells at low density activated EMT-like changes associated with increased expression of MT1-MMP [97, 98], while induction of EMT in MCF10A cells by exposure to TGFβ or expression of ErbB2 stimulated expression of MMP-2 and MMP-9, respectively [99, 100]. Decreased expression of singleminded-2s in mouse mammary epithelial cells or in MCF-7 breast cancer cells activated EMT and expression of MMP-2 [101], while expression of Snail in MCF-7 cells induced an MT1-MMP and MT2-MMP-dependent invasion program [102]. Activation of EMT in NMuMg cells by treatment with hydrogen peroxide led to activation of MMP-3, MMP-10, and MMP-13 [103], while induction of EMT by the Abl tyrosine kinase in the same cell line led to MMP-3 and MMP-9 expression [104]. As breast cancer progression is a complex process, it may be unsurprising that distinct profiles of MMPs are activated in systems that model different breast cancer stages and disease subtypes.
EMT of cancer cells may produce stromal-like derivatives that, while not intrinsically malignant, act to facilitate tumor progression through production of MMPs (Fig. 3c). Myofibroblasts are principal components of the reactive stroma surrounding breast cancers, and these cells have been found to have powerful tumor-promoting characteristics [105–108]. While myofibroblasts can be produced through activation of stromal fibroblasts or circulating fibrocytes, recent studies using mouse models have shown that myofibroblasts can be derived from epithelial cells by EMT [107, 109–113]. EMT functions in human breast cancer as well: stromal-like and myofibroblast-like cells surrounding breast tumors have been found to be derived from the epithelial cancer cells [107, 114]. It is further known that mammographic density, an established risk factor for and potential precursor of breast cancer [115, 116], is associated with fibroblast accumulation [117–122]. Studies of MMP localization in human tumors have shown that stromal fibroblasts are a major contributor to the production of many MMPs [123–125], and tumor progression and poor prognosis is associated with stromal expression of MMP-1, MMP-7, and MMP-12 [126], and with fibroblast-specific production of MMP-9, MMP-11, and MT1-MMP [124, 125]. Further defining which MMPs are produced by breast cancer-associated myofibroblasts, and how these MMPs act in tumor progression, will provide insight into how EMT-driven tumor progression can best be targeted therapeutically.
Unfortunately, there have been very few studies that assess histological correlates of EMT with expression of MMPs in human breast tissues. Studies with metaplastic breast carcinoma, a relatively uncommon subtype for which ongoing EMT processes are evident, have found that stromelysin-3/MMP-11 expression in epithelial cells is a prognostic factor for disease progression—patients who expressed more MMP-11 in epithelial carcinoma cells had significantly shorter disease-free survival [127]. More recently, profiling studies of metaplastic breast carcinoma have found that altered expression of MMPs and TIMPS were found in patients with more rapid disease progression [128]. However, a direct connection between MMPs and EMT that can be assessed by histological characteristics awaits future research.
Therapeutic Targeting of MMP-promoted EMT
An obvious point for intervention in MMP-induced or mediated EMT is the catalytic inhibition of MMPs themselves. Unfortunately, clinical trials of first- and second-generation small molecule MMP-inhibiting drugs in breast cancer and other cancers proved disappointing [129]. A phase III trial of the MMP inhibitor marimastat in patients with metastatic breast cancer found no therapeutic benefit [130], while phase II trials of marimastat and rebimastat in patients with early-stage breast cancer concluded that large adjuvant trials with these agents were not feasible due to musculoskeletal toxicity and failure to achieve therapeutic plasma levels [131, 132]. Many of the problems with the MMP inhibitors tested to date appear to stem in large part from a lack of specificity; the drugs employed simply target too many enzymes. This is a critical problem, because some MMPs appear to protect against tumor progression at certain stages of breast cancer development, and inhibition of these MMPs at the wrong time can lead to increased tumor aggressiveness [27, 133–135]. For example, high levels of MMP-8 have been shown to suppress breast cancer metastasis [136], potentially by increasing tumor cell adhesion to ECM and diminishing cellular invasive potential [137]; significantly, ribozyme-mediated knockdown of MMP-8 in a nonmetastatic, high MMP-8 breast cancer cell line conferred metastatic competence [136]. Thus, pharmacological inhibition of MMP-8 along with invasion- and metastasis-promoting MMPs would be anticipated to reduce or limit the potential benefit of the therapy.
As another consequence of poor specificity, clinical trials of MMP inhibitors were plagued by the serious side effect of musculoskeletal syndrome (MSS). This dose-limiting toxicity frequently resulted in failure to achieve targeted plasma levels, and in patients withdrawing from treatment, further compromising the statistical significance of trial outcomes [138]. The specific molecular target responsible for these side effects has not been conclusively identified; early candidates included MMP-1 and the ADAM family of metalloproteases, but synthetic inhibitors developed to minimize inhibition of these targets still produced MSS symptoms [138]. Remaining candidate mediators of MSS include MT1-MMP [135], metalloproteases outside of the MMP and ADAM families [139], or nonprotease metalloproteins [138]. To minimize off-target effects, well-tolerated MMP-directed therapeutics will need to achieve selectivity for the MMP family in preference to other metalloenzymes, as well as the ability to distinguish among MMPs.
The key challenges yet to be surmounted to bring MMP inhibitors to the clinic as an approach to combat MMP-mediated EMT and resulting cancer progression are (1) identification of the individual MMP targets implicated as primary drivers of EMT-promoted malignancy at specific points in tumor progression, and (2) development of therapeutic molecules capable of targeting these cancer-driving MMPs with exquisite selectivity. An attempt has already been made to synthesize existing data from current models into a master list of MMP drug targets versus “anti-targets”, for which pharmacological intervention would be presumed counterproductive [135]. However, the data currently available are insufficient to support definitive classification of most MMPs, particularly as questions remain regarding the extent to which various animal cancer models fully and faithfully reproduce the functional diversity of individual MMPs in cancer progression in humans. In particular, MMP-3, MMP-9, and MT1-MMP have all been suggested as drug anti-targets due to reports of antitumor effects associated with these MMPs in some model systems [135]; yet, the bulk of the literature supports the view that these are among the MMPs most directly implicated in promoting EMT, motility, invasion, and metastasis in cancer models. Rather than eliminating these MMPs from the drug target lineup, it would instead be prudent to cautiously pursue them, keeping in mind that further basic research into their functions in tumor development is necessary, and that their effective targeting for therapeutic benefit will require careful definition of the patient populations most likely to benefit, with regard to disease stage, pathological characteristics, and potential genetic modifiers. As an example of stage-specific considerations of potential importance in targeting MMP-induced EMT, it has been observed that MMP-3-induced EMT of breast epithelial cells is initially reversible upon withdrawal of the MMP, but eventually becomes permanent [31, 38], suggesting that therapeutic intervention with MMP inhibitors may be most effective at early stages of breast cancer development.
In the arena of more highly selective small molecule MMP inhibitors, slow progress is being made. These synthetic compounds typically feature a zinc-chelating group such as hydroxamate derivitized with peptidic or nonpeptidic groups designed to mimic a peptide substrate; they target the MMP active site zinc and substrate binding site [4, 140, 141]. Structure-based design of selective inhibitors has been hampered by the close structural homology of active sites and overlapping substrate specificities among the MMPs, and by the elastic and flexible nature of the MMP active site, which further complicates computational drug design even when high resolution crystal structures are available [141–144]. Current approaches to small molecule MMP inhibitors include optimization of compounds based on an array of different zinc-binding groups to yield more selective inhibitors toward a variety of MMPs [4, 145], as well as the development of non-zinc-binding inhibitors that selectively target unique aspects of the MMP-13 active site [145]. A less conventional approach has pursued development of irreversible mechanism-based inhibitors, selective for gelatinases MMP-2 and MMP-9, that covalently modify the catalytic glutamate residue of the MMP active site [145, 146]. In yet another approach, several groups have attempted to exploit the selective substrate binding exosites present on MMP accessory domains to develop selective allosteric inhibitors of MMPs; while a promising concept, this approach has yet to yield highly potent and selective drug leads [7].
An alternative to small molecule MMP inhibitors is presented by macromolecular protein therapeutics. Promising candidates for development include engineered variants of the natural MMP-inhibiting TIMPs, and MMP-targeting therapeutic antibodies. TIMPs offer the advantage of an extensive contact surface ideally evolved for high affinity interaction with MMP targets. Although the four native TIMPs possess only a limited ability to differentiate between the many members of the MMP family, mutational studies have established the potential for modulating binding specificity by alteration of key residues at the MMP-TIMP interface [147–152]. A recombinant triple mutant variant of the TIMP-1N-terminal domain, optimized for selectivity to MT1-MMP, was recently found to potently block MT1-MMP collagenase activity and CD44 shedding in breast cancer and fibrosarcoma cell culture models [153]. In another approach to selective MMP inhibition, several function blocking antibodies have been reported that selectively target individual MMPs [7]. In one recent and promising example, phage display technology was used to identify an MT1-MMP-selective human monoclonal antibody that blocked the proteolytic activity of the enzyme; this protein therapeutic was found to slow tumor progression and metastasis in an orthotopic xenograft model of breast cancer [154].
Thus, the challenges are clear: while some MMPs facilitate breast cancer development and could potentially be targeted for therapeutic benefit, others are essential for basic physiological processes, interference with which can have serious negative consequences. We need methods to target specific MMPs, as well as a much better understanding of which MMPs to target and when. Furthermore, while much has been learned about how to target the catalytic activities of MMPs, recent research has revealed that their noncatalytic accessory functions must also be considered. The efforts of chemists, biologists, bioengineers, and physicians must now be combined to discover selective drugs and reagents, to create the most informative experimental models in which to dissect the roles of MMPs in EMT-driven breast cancer progression, to develop and test optimal intervention strategies, and to effect the translation of these therapies into the clinic.
Abbreviations
- EMT:
-
epithelial-mesenchymal transition
- MMP:
-
matrix metalloproteinase
- ECM:
-
extracellular matrix
- TIMP:
-
tissue inhibitor of metalloproteinase
- PEX:
-
hemopexin
- TGF-β:
-
transforming growth factor-β
- LRP:
-
lipoprotein receptor-related protein
- WAP:
-
whey acidic protein
- MMTV:
-
mouse mammary tumor virus
- PyMT:
-
polyoma virus middle T-antigen
References
Quesada V, Ordonez GR, Sanchez LM, et al. The Degradome database: mammalian proteases and diseases of proteolysis. Nucleic Acids Res. 2009;37(Database issue):D239–43.
Tallant C, Marrero A, Gomis-Ruth FX. Matrix metalloproteinases: fold and function of their catalytic domains. Biochim Biophys Acta. 2009.
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–73.
Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006;94(7):941–6.
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–39.
Rosenblum G, Meroueh S, Toth M, et al. Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: challenging the cysteine switch dogma. J Am Chem Soc. 2007;129(44):13566–74.
Sela-Passwell N, Rosenblum G, Shoham T, et al. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim Biophys Acta. 2009.
Allan JA, Docherty AJ, Barker PJ, et al. Binding of gelatinases A and B to type-I collagen and other matrix components. Biochem J. 1995;309(Pt 1):299–306.
Bertini I, Fragai M, Luchinat C. Intra- and interdomain flexibility in matrix metalloproteinases: functional aspects and drug design. Curr Pharm Des. 2009;15(31):3592–605.
Piccard H, Van den Steen PE, Opdenakker G. Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J Leukoc Biol. 2007;81(4):870–92.
Chung L, Dinakarpandian D, Yoshida N, et al. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 2004;23(15):3020–30.
Lauer-Fields JL, Chalmers MJ, Busby SA, et al. Identification of specific hemopexin-like domain residues that facilitate matrix metalloproteinase collagenolytic activity. J Biol Chem. 2009;284(36):24017–24.
Gioia M, Monaco S, Fasciglione GF, et al. Characterization of the mechanisms by which gelatinase A, neutrophil collagenase, and membrane-type metalloproteinase MMP-14 recognize collagen I and enzymatically process the two alpha-chains. J Mol Biol. 2007;368(4):1101–13.
Tam EM, Moore TR, Butler GS, et al. Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem. 2004;279(41):43336–44.
Roeb E, Schleinkofer K, Kernebeck T, et al. The matrix metalloproteinase 9 (mmp-9) hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J Biol Chem. 2002;277(52):50326–32.
Monaco S, Gioia M, Rodriguez J, et al. Modulation of the proteolytic activity of matrix metalloproteinase-2 (gelatinase A) on fibrinogen. Biochem J. 2007;402(3):503–13.
McQuibban GA, Gong JH, Tam EM, et al. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–6.
McQuibban GA, Butler GS, Gong JH, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276(47):43503–8.
Overall CM, McQuibban GA, Clark-Lewis I. Discovery of chemokine substrates for matrix metalloproteinases by exosite scanning: a new tool for degradomics. Biol Chem. 2002;383(7–8):1059–66.
Ogata Y, Itoh Y, Nagase H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J Biol Chem. 1995;270(31):18506–11.
Goldberg GI, Strongin A, Collier IE, et al. Interaction of 92-kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. J Biol Chem. 1992;267(7):4583–91.
Ramos-DeSimone N, Hahn-Dantona E, Sipley J, et al. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274(19):13066–76.
Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–76.
Mori H, Tomari T, Koshikawa N, et al. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002;21(15):3949–59.
Kajita M, Itoh Y, Chiba T, et al. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol. 2001;153(5):893–904.
Mori H, Gjorevski N, Inman JL, et al. Self-organization of engineered epithelial tubules by differential cellular motility. Proc Natl Acad Sci U S A. 2009;106(35):14890–5.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74.
Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–33.
Boire A, Covic L, Agarwal A, et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120(3):303–13.
Noe V, Fingleton B, Jacobs K, et al. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114(Pt 1):111–8.
Lochter A, Galosy S, Muschler J, et al. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139(7):1861–72.
Deryugina EI, Ratnikov BI, Postnova TI, et al. Processing of integrin alpha(v) subunit by membrane type 1 matrix metalloproteinase stimulates migration of breast carcinoma cells on vitronectin and enhances tyrosine phosphorylation of focal adhesion kinase. J Biol Chem. 2002;277(12):9749–56.
Ratnikov BI, Rozanov DV, Postnova TI, et al. An alternative processing of integrin alpha(v) subunit in tumor cells by membrane type-1 matrix metalloproteinase. J Biol Chem. 2002;277(9):7377–85.
Ribeiro AS, Albergaria A, Sousa B, et al. Extracellular cleavage and shedding of P-cadherin: a mechanism underlying the invasive behaviour of breast cancer cells. Oncogene. 2010;29(3):392–402.
Strongin AY, Collier I, Bannikov G, et al. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270(10):5331–8.
Van den Steen PE, Proost P, Wuyts A, et al. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96(8):2673–81.
Almholt K, Green KA, Juncker-Jensen A, et al. Extracellular proteolysis in transgenic mouse models of breast cancer. J Mammary Gland Biol Neoplasia. 2007;12(1):83–97.
Sternlicht MD, Lochter A, Sympson CJ, et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98(2):137–46.
Rudolph-Owen LA, Chan R, Muller WJ, et al. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998;58(23):5500–6.
Ha HY, Moon HB, Nam MS, et al. Overexpression of membrane-type matrix metalloproteinase-1 gene induces mammary gland abnormalities and adenocarcinoma in transgenic mice. Cancer Res. 2001;61(3):984–90.
Masson R, Lefebvre O, Noel A, et al. In vivo evidence that the stromelysin-3 metalloproteinase contributes in a paracrine manner to epithelial cell malignancy. J Cell Biol. 1998;140(6):1535–41.
Andarawewa KL, Boulay A, Masson R, et al. Dual stromelysin-3 function during natural mouse mammary tumor virus-ras tumor progression. Cancer Res. 2003;63(18):5844–9.
Matrisian LM. Cancer biology: extracellular proteinases in malignancy. Curr Biol. 1999;9(20):R776–8.
Martin MD, Carter KJ, Jean-Philippe SR, et al. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008;68(15):6251–9.
Szabova L, Chrysovergis K, Yamada SS, et al. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene. 2008;27(23):3274–81.
Thiolloy S, Halpern J, Holt GE, et al. Osteoclast-derived matrix metalloproteinase-7, but not matrix metalloproteinase-9, contributes to tumor-induced osteolysis. Cancer Res. 2009;69(16):6747–55.
Witty JP, Lempka T, Coffey Jr RJ, et al. Decreased tumor formation in 7, 12-dimethylbenzanthracene-treated stromelysin-1 transgenic mice is associated with alterations in mammary epithelial cell apoptosis. Cancer Res. 1995;55(7):1401–6.
Radisky DC. Epithelial-mesenchymal transition. J Cell Sci. 2005;118(Pt 19):4325–6.
Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120(11):1351–83.
Thiery JP, Acloque H, Huang RY, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Hugo H, Ackland ML, Blick T, et al. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213(2):374–83.
Duband JL. Neural crest delamination and migration: integrating regulations of cell interactions, locomotion, survival and fate. Adv Exp Med Biol. 2006;589:45–77.
Duong TD, Erickson CA. MMP-2 plays an essential role in producing epithelial-mesenchymal transformations in the avian embryo. Dev Dyn. 2004;229(1):42–53.
Cai DH, Brauer PR. Synthetic matrix metalloproteinase inhibitor decreases early cardiac neural crest migration in chicken embryos. Dev Dyn. 2002;224(4):441–9.
Cai DH, Vollberg Sr TM, Hahn-Dantona E, et al. MMP-2 expression during early avian cardiac and neural crest morphogenesis. Anat Rec. 2000;259(2):168–79.
Cantemir V, Cai DH, Reedy MV, et al. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression during cardiac neural crest cell migration and its role in proMMP-2 activation. Dev Dyn. 2004;231(4):709–19.
Runyan RB, Heimark RL, Camenisch TD, et al. Epithelial-Mesenchymal Transformation in the Embryonic Heart. In: Savagner P, editors. Rise and Fall of Epithelial Phenotype: Springer US; 2005. p. 40–55.
Song W, Jackson K, McGuire PG. Degradation of type IV collagen by matrix metalloproteinases is an important step in the epithelial-mesenchymal transformation of the endocardial cushions. Dev Biol. 2000;227(2):606–17.
Alexander SM, Jackson KJ, Bushnell KM, et al. Spatial and temporal expression of the 72-kDa type IV collagenase (MMP-2) correlates with development and differentiation of valves in the embryonic avian heart. Dev Dyn. 1997;209(3):261–8.
Rupp PA, Visconti RP, Czirok A, et al. Matrix metalloproteinase 2-integrin alpha(v)beta3 binding is required for mesenchymal cell invasive activity but not epithelial locomotion: a computational time-lapse study. Mol Biol Cell. 2008;19(12):5529–40.
Fata JE, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004;6(1):1–11.
Sternlicht MD, Kouros-Mehr H, Lu P, et al. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74(7):365–81.
Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296(5570):1046–9.
Kouros-Mehr H, Werb Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 2006;235(12):3404–12.
Wiseman BS, Sternlicht MD, Lund LR, et al. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162(6):1123–33.
Sympson CJ, Talhouk RS, Alexander CM, et al. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125(3):681–93.
Wang YA, Shen K, Wang Y, et al. Retinoic acid signaling is required for proper morphogenesis of mammary gland. Dev Dyn. 2005;234(4):892–9.
Hirai Y, Lochter A, Galosy S, et al. Epimorphin functions as a key morphoregulator for mammary epithelial cells. J Cell Biol. 1998;140(1):159–69.
Hirai Y, Radisky D, Boudreau R, et al. Epimorphin mediates mammary luminal morphogenesis through control of C/EBPbeta. J Cell Biol. 2001;153(4):785–94.
Radisky DC, Hirai Y, Bissell MJ. Delivering the message: epimorphin and mammary epithelial morphogenesis. Trends Cell Biol. 2003;13(8):426–34.
Chen CS, Nelson CM, Khauv D, et al. Homology with vesicle fusion mediator syntaxin-1a predicts determinants of epimorphin/syntaxin-2 function in mammary epithelial morphogenesis. J Biol Chem. 2009;284(11):6877–84.
Xian W, Schwertfeger KL, Vargo-Gogola T, et al. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J Cell Biol. 2005;171(4):663–73.
Cheng S, Lovett DH. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am J Pathol. 2003;162(6):1937–49.
Cheng S, Pollock AS, Mahimkar R, et al. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J. 2006;20(11):1898–900.
Tan TK, Zheng G, Hsu TT, et al. Macrophage Matrix Metalloproteinase-9 Mediates Epithelial-Mesenchymal Transition in Vitro in Murine Renal Tubular Cells. Am J Pathol. 2010.
Zheng G, Lyons JG, Tan TK, et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol. 2009;175(2):580–91.
Cowden Dahl KD, Symowicz J, Ning Y, et al. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008;68(12):4606–13.
West-Mays JA, Pino G. Matrix metalloproteinases as mediators of primary and secondary cataracts. Expert Rev Ophthalmol. 2007;2(6):931–8.
Illman SA, Lehti K, Keski-Oja J, et al. Epilysin (MMP-28) induces TGF-beta mediated epithelial to mesenchymal transition in lung carcinoma cells. J Cell Sci. 2006;119(Pt 18):3856–65.
Cao J, Chiarelli C, Richman O, et al. Membrane type 1 matrix metalloproteinase induces epithelial-to-mesenchymal transition in prostate cancer. J Biol Chem. 2008;283(10):6232–40.
Sternlicht MD, Bissell MJ, Werb Z. The matrix metalloproteinase stromelysin-1 acts as a natural mammary tumor promoter. Oncogene. 2000;19:1102–13.
Sympson CJ, Bissell MJ, Werb Z. Mammary gland tumor formation in transgenic mice overexpressing stromelysin-1. Semin Cancer Biol. 1995;6(3):159–63.
Lochter A, Srebrow A, Sympson CJ, et al. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J Biol Chem. 1997;272(8):5007–15.
Radisky DC, Levy DD, Littlepage LE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436(7047):123–7.
Fiegen D, Haeusler LC, Blumenstein L, et al. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004;279(6):4743–9.
Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem. 2003;278(50):50442–8.
Singh A, Karnoub AE, Palmby TR, et al. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23(58):9369–80.
Jordan P, Brazao R, Boavida MG, et al. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene. 1999;18(48):6835–9.
Schnelzer A, Prechtel D, Knaus U, et al. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19(26):3013–20.
Nelson CM, Khauv D, Bissell MJ, et al. Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. J Cell Biochem. 2008.
Gilles C, Newgreen DF, Sato H, et al. Matrix Metalloproteinases and Epithelial-to-Mesenchymal Transition: Implications for Carcinoma Metastasis. In: Savagner P, editor. Rise and Fall of Epithelial Phenotype: Springer US; 2005. p. 297–315.
Gilles C, Polette M, Seiki M, et al. Implication of collagen type I-induced membrane-type 1-matrix metalloproteinase expression and matrix metalloproteinase-2 activation in the metastatic progression of breast carcinoma. Lab Invest. 1997;76(5):651–60.
Gilles C, Polette M, Birembaut P, et al. Expression of c-ets-1 mRNA is associated with an invasive, EMT-derived phenotype in breast carcinoma cell lines. Clin Exp Metastasis. 1997;15(5):519–26.
Martorana AM, Zheng G, Crowe TC, et al. Epithelial cells up-regulate matrix metalloproteinases in cells within the same mammary carcinoma that have undergone an epithelial-mesenchymal transition. Cancer Res. 1998;58(21):4970–9.
Janda E, Lehmann K, Killisch I, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156(2):299–313.
Jechlinger M, Grunert S, Tamir IH, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22(46):7155–69.
Gilles C, Polette M, Coraux C, et al. Contribution of MT1-MMP and of human laminin-5 gamma2 chain degradation to mammary epithelial cell migration. J Cell Sci. 2001;114(Pt 16):2967–76.
Sarrio D, Rodriguez-Pinilla SM, Hardisson D, et al. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989–97.
Kim ES, Sohn YW, Moon A. TGF-beta-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007;252(1):147–56.
Kim IY, Yong HY, Kang KW, et al. Overexpression of ErbB2 induces invasion of MCF10A human breast epithelial cells via MMP-9. Cancer Lett. 2009;275(2):227–33.
Laffin B, Wellberg E, Kwak HI, et al. Loss of singleminded-2s in the mouse mammary gland induces an epithelial-mesenchymal transition associated with up-regulation of slug and matrix metalloprotease 2. Mol Cell Biol. 2008;28(6):1936–46.
Ota I, Li XY, Hu Y, et al. Induction of a MT1-MMP and MT2-MMP-dependent basement membrane transmigration program in cancer cells by Snail1. Proc Natl Acad Sci U S A. 2009;106(48):20318–23.
Mori K, Shibanuma M, Nose K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004;64(20):7464–72.
Allington TM, Galliher-Beckley AJ, Schiemann WP. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009;23(12):4231–43.
Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101(4):830–9.
Faouzi S, Le Bail B, Neaud V, et al. Myofibroblasts are responsible for collagen synthesis in the stroma of human hepatocellular carcinoma: an in vivo and in vitro study. J Hepatol. 1999;30(2):275–84.
Petersen OW, Nielsen HL, Gudjonsson T, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162(2):391–402.
Ronnov-Jessen L, Petersen OW, Koteliansky VE, et al. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95(2):859–73.
Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006.
Lee EH, Joo CK. Role of transforming growth factor-beta in transdifferentiation and fibrosis of lens epithelial cells. Invest Ophthalmol Vis Sci. 1999;40(9):2025–32.
Li JH, Wang W, Huang XR, et al. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol. 2004;164(4):1389–97.
Nightingale J, Patel S, Suzuki N, et al. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J Am Soc Nephrol. 2004;15(1):21–32.
Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3(4):377–82.
Moinfar F, Man YG, Arnould L, et al. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60(9):2562–6.
Boyd NF, Rommens JM, Vogt K, et al. Mammographic breast density as an intermediate phenotype for breast cancer. Lancet Oncol. 2005;6(10):798–808.
Kelemen LE, Sellers TA, Vachon CM. Can genes for mammographic density inform cancer aetiology? Nat Rev Cancer. 2008;8(10):812–23.
Bartow SA, Pathak DR, Mettler FA. Radiographic microcalcification and parenchymal patterns as indicators of histologic “high-risk” benign breast disease. Cancer. 1990;66(8):1721–5.
Boyd NF, Jensen HM, Cooke G, et al. Mammographic densities and the prevalence and incidence of histological types of benign breast disease. Reference Pathologists of the Canadian National Breast Screening Study. Eur J Cancer Prev. 2000;9(1):15–24.
Bright RA, Morrison AS, Brisson J, et al. Relationship between mammographic and histologic features of breast tissue in women with benign biopsies. Cancer. 1988;61(2):266–71.
Buchanan JB, Weisberg BF, Sandoz JP, et al. Selected prognostic variables for mammographic parenchymal patterns. Cancer. 1981;47(9):2135–7.
Urbanski S, Jensen HM, Cooke G, et al. The association of histological and radiological indicators of breast cancer risk. Br J Cancer. 1988;58(4):474–9.
Wellings SR, Wolfe JN. Correlative studies of the histological and radiographic appearance of the breast parenchyma. Radiology. 1978;129(2):299–306.
Heppner KJ, Matrisian LM, Jensen RA, et al. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149(1):273–82.
Del Casar JM, Gonzalez LO, Alvarez E, et al. Comparative analysis and clinical value of the expression of metalloproteases and their inhibitors by intratumor stromal fibroblasts and those at the invasive front of breast carcinomas. Breast Cancer Res Treat. 2009;116(1):39–52.
Vizoso FJ, Gonzalez LO, Corte MD, et al. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br J Cancer. 2007;96(6):903–11.
Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27.
Ahmad A, Hanby A, Dublin E, et al. Stromelysin 3: an independent prognostic factor for relapse-free survival in node-positive breast cancer and demonstration of novel breast carcinoma cell expression. Am J Pathol. 1998;152(3):721–8.
Lien HC, Hsiao YH, Lin YS, et al. Molecular signatures of metaplastic carcinoma of the breast by large-scale transcriptional profiling: identification of genes potentially related to epithelial-mesenchymal transition. Oncogene. 2007;26(57):7859–71.
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–92.
Sparano JA, Bernardo P, Stephenson P, et al. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol. 2004;22(23):4683–90.
Miller KD, Gradishar W, Schuchter L, et al. A randomized phase II pilot trial of adjuvant marimastat in patients with early-stage breast cancer. Ann Oncol. 2002;13(8):1220–4.
Miller KD, Saphner TJ, Waterhouse DM, et al. A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin Cancer Res. 2004;10(6):1971–5.
Fingleton B. Matrix metalloproteinases: roles in cancer and metastasis. Front Biosci. 2006;11:479–91.
Martin MD, Matrisian LM. The other side of MMPs: protective roles in tumor progression. Cancer Metastasis Rev. 2007;26(3–4):717–24.
Overall CM, Kleifeld O. Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–39.
Montel V, Kleeman J, Agarwal D, et al. Altered metastatic behavior of human breast cancer cells after experimental manipulation of matrix metalloproteinase 8 gene expression. Cancer Res. 2004;64(5):1687–94.
Gutierrez-Fernandez A, Fueyo A, Folgueras AR, et al. Matrix metalloproteinase-8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res. 2008;68(8):2755–63.
Fingleton B. MMPs as therapeutic targets-still a viable option? Semin Cell Dev Biol. 2008;19(1):61–8.
Saghatelian A, Jessani N, Joseph A, et al. Activity-based probes for the proteomic profiling of metalloproteases. Proc Natl Acad Sci U S A. 2004;101(27):10000–5.
Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006;25(1):115–36.
Rao BG. Recent developments in the design of specific Matrix Metalloproteinase inhibitors aided by structural and computational studies. Curr Pharm Des. 2005;11(3):295–322.
Bertini I, Calderone V, Cosenza M, et al. Conformational variability of matrix metalloproteinases: beyond a single 3D structure. Proc Natl Acad Sci U S A. 2005;102(15):5334–9.
Matter H, Schudok M. Recent advances in the design of matrix metalloprotease inhibitors. Curr Opin Drug Discov Devel. 2004;7(4):513–35.
Moy FJ, Chanda PK, Chen J, et al. Impact of mobility on structure-based drug design for the MMPs. J Am Chem Soc. 2002;124(43):12658–9.
Jacobsen JA, Major Jourden JL, Miller MT, et al. To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta. 2010;1803(1):72–94.
Ikejiri M, Bernardo MM, Meroueh SO, et al. Design, synthesis, and evaluation of a mechanism-based inhibitor for gelatinase A. J Org Chem. 2005;70(14):5709–12.
Lee MH, Rapti M, Knauper V, et al. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J Biol Chem. 2004;279(17):17562–9.
Lee MH, Rapti M, Murphy G. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J Biol Chem. 2003;278(41):40224–30.
Meng Q, Malinovskii V, Huang W, et al. Residue 2 of TIMP-1 is a major determinant of affinity and specificity for matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1’ residue of substrate. J Biol Chem. 1999;274(15):10184–9.
Nagase H, Brew K. Designing TIMP (tissue inhibitor of metalloproteinases) variants that are selective metalloproteinase inhibitors. Biochem Soc Symp 2003;(70):201–12.
Wei S, Chen Y, Chung L, et al. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J Biol Chem. 2003;278(11):9831–4.
Williamson RA, Hutton M, Vogt G, et al. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J Biol Chem. 2001;276(35):32966–70.
Lee MH, Atkinson S, Rapti M, et al. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)-1 against native membrane type 1 matrix metalloproteinase (MT1-MMP) in a cell-based environment. Cancer Lett. 2009.
Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69(4):1517–26.
DeLano WL. The PyMOL Molecular Graphics System. In. San Carlos, CA, USA: DeLano Scientific. 2002.
Morgunova E, Tuuttila A, Bergmann U, et al. Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc Natl Acad Sci U S A. 2002;99(11):7414–9.
Financial Support
This work is supported by grants from the National Cancer Institute (CA122086, CA128660, and CA132879 to DCR), from the James and Esther King Foundation (07KN09 to DCR; 08KN12 to ESR), from the Bankhead-Coley Foundation (09BB17 to ESR), from the Susan B. Komen foundation (FAS0703855 to DCR), and by the Mayo Clinic Breast Cancer Specialized Program of Research Excellence (SPORE) grant CA116201 (PI James Ingle MD) from the National Institutes of Health.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Radisky, E.S., Radisky, D.C. Matrix Metalloproteinase-Induced Epithelial-Mesenchymal Transition in Breast Cancer. J Mammary Gland Biol Neoplasia 15, 201–212 (2010). https://doi.org/10.1007/s10911-010-9177-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-010-9177-x