
Abstract
The buffering Cadmium–Imidazole Coordination Polymer (Cd–Im-CP) was synthesized hydrothermally from cadmium chloride and imidazole at 70 °C and then was subjected to doping- by the non-metal sulfur using Na2S solution as a novel modification strategy to produce S–Cd–Im CPs. To investigate doping nature and its effects, Cd–Im CP and S–Cd–Im CPs were characterized applying different analyses techniques, FTIR, Raman, PXRD, SEM/EDX, TGA, and UV–Vis DRS analyses. Characterizations showed the successful chemical doping of sulfur. The inclusion of sulfur within chemical CP structure caused narrowing of material’s bandgap from 4.55 and 3.4 eV to 4.25 and 2.35 eV for S–Cd–Im CPs allowing it for photoresponse towards Visible-light. Both Cd–Im CP and S–Cd–Im CPs were applied for photocatalytic degradation of the selected dyes methylene blue (MB),and methyl orange (MO) employing visible and UV irradiations considering three different initial pH levels to investigate the consequence of sulfur doping. After eliminating the photolysis effect, the best degradation by S–Cd–Im CPs was recorded for MB at initial pH 4 being 13 fold that is for Cd–Im CP. The highest apparent turnover frequencies are 1.2 × 10−3 h−1 for MB at initial pH 10 and 1.03 × 10−4 h−1 for MO at initial pH 4 are given by 10S–Cd–IM CP under Visible-light. Generally, S–Cd–Im CPs remarkably improved photocatalysis degradation of both the dyes for all initial pH levels under Visible-light.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Coordination polymers (CPs) and metal organic frameworks (MOFs) are hybrid materials composed of metal nodes and organic linkers connected via strong coordination bondings to construct porous 1D, 2D, and 3D dimensional networks. Most of these materials exhibit remarkable large surface area giving the grand ability for remediating wastewater via adsorption process. However, when being photoactive, i.e. responsive to the applied light, these materials can be pertained as photocatalysts for wastewater remediation. Such photocatalysis is an efficient approach since partial degradations or even complete mineralization of organic pollutants can be achieved [1]
Over the past two decades, CPs and MOFs and Metal oxides composties have elucidated their potential for heterogeneous photocatalytic degradation of dyes/organics in waters [2,3,4,5,6,7,8,9,10,11]. Until 2014, most of the studied photoactive CPs/MOFs for dyes degradation have been utilized under UV or UV–Vis light irradiation. On the other hand, a relatively small number of CPs/MOFs have been utilized under Visible-light where H2O2, as an auxiliary oxidant, usually was a need to assist degradation [12].
In general, to be effective, MOFs and CPs should have semiconduction behavior when exposed to light irradiation. The organic component, the linker, can act as an antenna to absorb light, natural or artificial, with subsequent activation of the metal nodes via ligand to metal charge transition (LMCT). On a whole structural basis, typically, light absorption promotes an electron to a higher energy level which then migrate to the electron acceptor moiety within the structure where the resulting negative charges (electrons) and positive charges (holes) can be used to drive chemical reactions [13,14,15,16]
Hence, mechanism of action can be regarded as excitation of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) when MOF/CP is irradiated with light, thus leaving a hole in the HOMO. In an aqueous medium, the holes can interact with OH, forming the ˙OH radicals which oxidizes the organic compounds [17]. On the other hand, the excited electrons (e−) can interact with dissolved oxygen to produce oxygen radicals which in turn transform to ˙OH radicals. Hence, the photocatalytic performance of the photoactive MOF involves the generation of electron–hole pairs in the conduction and valence bands of the MOF/CP respectively. Thus, an important criterion of MOFs/CPs as photoactive materials for photocatalytic applications is the ability to absorb or harvest light energy and conduit it into chemical reaction [18].
Some recent progresses in CPs-based and MOFs-based photocatalysis for wastewater remediation under Visible-light considered some strategies so as to modify or regulate the pristine CPs/MOFs for more efficient photocatalytic performances. These strategies include, among others, ligand functionalization, metal-ion immobilization, dye sensitization, loading of metal-nanoparticle, decoration by carbon material and conventional semiconductor coupling. Such strategies may result in, to one degree or another, the extension of light absorption toward Visible-light band, efficient generation of electron–hole pairs, valuable separation and transfer of photogenerated charges, as well as good recyclability [12]. The outstanding feature of porosity of such hybrid materials candidates the chemical structure of their channel walls for post-synthetic modification of the organic linkers by incorporating new organic functionalities or introducing metal cations and hence could enable/improve catalysis [19].
By principle, photoactive CPs/MOFs have some distinctive features and the most important are: (i) the tunable active sites (such as metal oxygen clusters and organic linkers) in MOFs/CPs enable efficient utilization of solar energy, and (ii) the topological structure of MOFs/CPs crafts the active sites relatively dispersed resulting in high stability and effectiveness [1].
In 2007, Alvaro et al.elucidated for the first time that MOF-5 can be used as photocatalysts to degrade phenol in water under UV-light irradiation. The terephthalate organic linker of the MOF, when in solution, tends to generate some charges and it is suggested that MOF-5 has a semiconducting property. Since then, several inorganic/organic hybrid materials have been studied as photocatalysts for the degradation of organic pollutants [1, 20]. For example, some photoactive MOFs containing Fe, Cr, Zr, and Ti metal ions show suitable stability in water and can absorb and convert light energy. These MOFs usually possess a suitable bandgap which enables Visible-light excitation; hence, they are considered as good candidates for photocatalytic degradations of organic pollutants [18, 21, 22].
As a traditional background, it is known that incorporating transition-metal ions into the lattice of TiO2 introduces additional energy levels within the bandgap. These levels help extending/shifting absorption of light from the UV to the visible region, thus enhancing the material’s photoresponse [23]. Hence lately and similar to classical semiconductors, CPs and MOFs have been themed to modification by doping, specifically by metals, for more efficient photocatalytic performance.
Wang et al.presented a modification of UiO-66 MOF by titanium where optical properties have been enhanced due to the formation of oxo-bridged hetero-Zr–Ti clusters. XRD analysis study disclosed that titanium species were not simply grafted or encaged in the UiO-66 framework; however instead, Ti species might be cooperated with Zr to form oxo-bridged hetero-Zr–Ti clusters, which then act as the skeleton of the UiO-66(Ti). Such structural modification impacted physically the material. The UV–Vis absorption edges of the as-prepared UiO-66(Ti) nanocomposites showed a shift to longer wavelengths compared with the original UiO-66. This red-shift of UV–visible absorption edge implies the decrease in the bandgap after the introduction of titanium species. Consequently the modified sample, UiO-66(1.25Ti), has shown improved MB removal efficiency of 87.1% under simulated sun-light irradiation [24]. Li et al.have mechanochemically introduced active Ni metal centers into ZIFs. One sample, BIT-11b with photoactive Ni centers and open channels, has exhibited an outstanding photocatalytic activity towards degrading MB under Visible-light within 25 min. It was suggested that the excited electron from Ni (with variable valence) is captured by electronegative substances such as molecular oxygen in solution, which would transform into highly active peroxide anions and thereafter achieve further oxidation/degradation of MB [25]. Reda et al.have post-synthetically modified NH2-MIL-125 MOF with Cr(III), acting as hole acceptors, in a form complex. This modification decreased the bandgap from 2.6 to 2.21 eV whose consequence was the increase of MB degradation under Visible-light. Also in the same work, it is shown that NH2-MIL-125 photocatalytic activity has been much improved by doping with Ag nanoparticles, formed in situ by the reduction of Ag(I). The resulting modified MOF has a bandgap of 2.09 eV which is suitable for Visible-light absorption. It is claimed that the Ag nanoparticles are able to accept the MOF’s photo-generated electrons, thus avoiding electron–hole recombination [23]. Botas et al.showed that exchanging Zn atoms in pure MOF-74 with varying amounts of Mg, Cu, or Co in the inorganic cluster the modified the bandgap in the range of 1.88–2.88 eV [26]. Most of these modifications renovate pristine samples being responsive to Visible-light and/or being of less electron/hole recombination, however by applying metal doping in most cases.
On the other hand, however, researches pertaining the modification of CPs and MOFs by non-metals are very seldom; even though this modification has shown enhanced photocatalytic activity [27, 28]. Indeed, many classical semiconductors, such as TiO2, have been modified with non-metals (such as N, S, O, F and C) and have shown considerably enhanced performances considering photocatalysis [29,30,31]. Photocatalysis enhancement by non-metal dopants is primarily referred to, among others, the formation of intermediate energy levels which modulates the bandgap and/or act as trapping center for the photo-generated electrons that decreases the recombination rate of charge carriers [31].
Very recently, we have presented the solid buffering Cd–Im CP (based on the d10 Cd(II) and imidazole) as a promising photocatalyst for degrading cationic and anionic dyes. Cd–Im CP has shown appropriate degrading performance towards MB and MO [32,33,34]. In the present work, Cd–Im CP has been synthesized and then post-modified with sulfur as a non-metal dopant. The intended selection of sulfur to dope by is based on the ease of proposed Lewis acid–base Cd–S adduct formation. Hence, the strategy is to modulate the metal node. Cd–Im CP and sulfur modified samples, S–Cd–Im CPs, were comparatively characterized to explore the nature of sulfur modification. Then, pristine and modified CPs were evaluated for their photocatalytic capability for degrading the cationic MB and the anionic MO under visible-light and UV-light. Doping CPs by non-metals should effectually enhances photocatalysis performance due to expected strong interaction within the CP matrix. This work presents the correlation of degradation profiles of these dyes with the applied conditions, the light type and the applied initial pH, to facilitate investigating the effect and the role of doped sulfur in enhancing degradation. Sulfur doped samples, S–Cd–Im CPs, showed improved MB degradation for all initial pH levels under Visible-light. Generally, it has been found that non-metal doping could narrow the original bandgap and/or reduce e−/h+ recombination [35]. This work should be the first, to the best of our knowledge, to introduce some plausible mechanistic elucidations of degradation concerning non-metal doping under the observance of buffering effect.
2 Experimental
2.1 Hydrothermal Synthesis of Pristine and Sulfur Modified Cd–Im CPs
Cd–Im CP was prepared according to our previously reported method [32]. Briefly, ammoniated imidazole solution (136 mg (2 mM)/10 ml DI water) was added to the CdCl2.2H2O solution (219 mg (1 mM)/10 ml DI water) producing an immediate slim turbid mix. The mix was heated in an oven at 70 °C for about 2 h during which Cd–Im CP powder precipitated progressively. The powder was collected and washed several times using warm DI water and lastly was dried at 80 °C, and then stored for the latter use: sulfur-modification, characterizations and photocatalytic degradation experiments.
S-modified samples were prepared by treating dried pristine Cd–Im CP by two Na2S solutions of different concentrations, 5 and 10 ppm. Treatment was performed by soaking pristine samples (100 mg) each in 20 ml of Na2S solution of mentioned concentrations for 24 h at room temperature. After treatment, samples (5S–Cd–Im CP and 10S–Cd–Im CP) were collected, washed several times and then dried and stored for later characterizations and photocatalytic degradation experiments.
2.2 Characterization of Cd–Im CPs and S–Cd–Im CPs
The chemical structure of Cd–Im CP and S–Cd–Im CPs were investigated by FTIR (standard KBr pellet method, recording range 400 to 3500 cm−1, 100 scans, and resolution of 4 cm−1, Jasco FT/IR 4100). In addition, Raman spectroscopy (Sentera, Bruker, Germany, with a 785 nm, 100 mW excitation laser) was also applied for confirming the changes and the presence of functional groups. The crystalline nature of the CPs was studied by PXRD (Shimadzu XD-l, with Cu-Kα radiation). Surface morphologies of CPs were determined using SEM (Zeiss EVO-10 microscopy) with making use of the coupled EDX to explore the elemental compositions of samples. UV–VIS DRS equipped with the integrating sphere accessory for diffuse reflectance (Jasco V 530 spectrometer, Japan) was applied and spectra were recorded for all samples. Derived from these spectra, the bandgap values (Eg) were calculated from Tauc’s plot, (F.(hν))0.5 versus hν where F = (1 − R)/(2R), R is reflectance, h is Plank’s constant, and ν is the frequency.
For thermal stability investigation, TGA measurements were applied for all CPs (TGA discovery, TA instruments, USA) where 1 mg of each CP was heated at 20 °C/min from about 27 °C up to about 700 °C under N2 atmosphere (30 ml/min). CPs stability in aqueous media is an important criterion regarding photocatalytic degradation applications in water. Many CPs are not that stable in aqueous media and therefore are not suitable for water treatment. Two tests were performed to evaluate the stability of Cd–Im CP: mass loss and FTIR analysis. For the mass loss test, an amount of the as-synthesized Cd–Im CP was washed thoroughly by 7 batches of warm DI water and was dried at 80 °C. A dried amount sample of (100 mg) was soaked in DI water (100 ml) for 24 h at room temperature. Then, the sample was separated, dried again and weighed to determine the mass loss percentage.
2.3 Photocatalysis Measurements
The photocatalytic activity of CPs was evaluated by considering the degradation of two different types of dyes, the cationic MB and the anionic MO, considering solutions of different initial pH levels 4, 7, and 10 at room temperature. Irradiation was implemented using a Visible-lamp (35 W, λ = 400–800 nm) and UV-lamp (25 W, 254 nm) separately. In a typical photocatalysis experiment, a CP sample (50 mg, thoroughly prewashed/dried) was added into a beaker (100 ml) containing an aqueous solution of MB or MO (25 ml, 5 ppm). Solutions were imported into a simple shaking setup (100 rpm) equipped with top-source light from a specified distance. Periodically, a sample (3 ml) was withdrawn from photocatalysis solution to determine the remnant concentration of the dye using Agilent 60 UV–Vis spectrophotometer (Jasco V 530 spectrometer, Japan) at 664 and 465 nm for MB and MO respectively. Corresponding photolysis experiments were also carried out under the same conditions for comparison. To evaluate degradation performance, degradation efficiency is presented as D% = 100(Co–Ct)/Co, where Co and Ct are respectively the initial concentration and remnant concentration at a certain time t.
3 Results and Discussion
3.1 Characterization of Cd–Im CP and S–Cd–Im CPs
3.1.1 FTIR and Raman Analyses
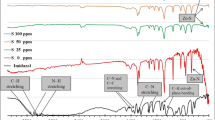
Figure 1 shows the IR spectra of imidazole, Cd–Im CP and S–Cd–Im CPs. As mentioned earlier, Cd–Im CP has been previously synthesized and its IR analysis compared to that of imidazole was discussed and assigned the creation of Cd–Im CP [32]. In brief, the spectrum of imidazole shows its characteristic peaks for C–H stretching vibrations at 3135 and 2900 cm−1. These peaks also appeared in the Cd–Im CP spectrum with some shift. This confirms the intactness of imidazole structure within Cd–Im CP matrix. The N–H stretching vibrations shows characteristic peak located at 2838 cm−1 for both imidazole and Cd–Im CP. Peaks at 1457 and 1365 cm−1 are designated to C–N stretching vibrational modes and these peaks have been shifted to 1380 and 1480 cm−1 in Cd–Im CP so the structure of imidazole is still found in the Cd–Im CP. The stretching modes of C=N and C=C are observed at 1550 and 1527 cm−1 for imidazole while they are observed at 1593 and 1630 cm−1 for Cd–Im CP, i.e. slight shift is recorded. This shift to the higher energy band suggests more rigidity in the structure attributed to the Cd-coordination.

FTIR spectra of imidazole, Cd–Im CP, and S–Cd–Im CPs
Herein, the effect of the sulfur modification is purposely presented. The imidazole spectrum illustrates its known characteristic peaks. These peaks also have appeared in the S–Cd–Im CPs spectra confirming that imidazole structure is still present in these CPs. All peaks of the S–Cd–Im CPs are approximately the same as that of the pristine Cd–Im CP which indicates the conservation of the main structure. However, the appearance of new peaks at 435 cm−1 in the new S–Cd–Im CPs should indicate the presence of Cd−S bond in the modified CPs. Prominently, strong triplet peaks appear in the range from 3500 to 3700 cm−1 and could represent the new attached –SH group to Cd. These peaks inevitably characterize S-bonding within S–Cd–Im CPs samples and indicate, as anticipated, the successful sulfur chemical inclusion in pristine Cd–Im CP structure. The successful inclusion of sulfur is also supported by SEM/EDX and XRD analyses as will be shown later. More interesting, the broadband appearing at about 3400 cm−1 for pristine Cd–Im CP, assigned to the OH-stretching mode, has been replaced by the mentioned triplet of –SH for S–Cd–Im CPs [36].
Raman analysis of Cd–Im CP was also presented compared to imidazole in the previous work which confirmed the successful coordination of Cd with imidazole forming Cd–Im CP [32]. Herein also, the consequence of sulfur modification is considered. Raman spectra of imidazole, Cd–Im CP and S–Cd–Im CPs are shown in Fig. 2. Generally, spectra of S–Cd–Im CPs are very similar to that of Cd–Im CP indicating that sulfur modification did not alter appreciably the pristine structure. The first region, 50–250 cm−1, shows that sulfur modification did not affect the whole lattice vibrations preserving their crystalline nature. The second region that of fingerprint, 250–1800 cm−1, indicates that sulfur modification has no effect on functional vibration energies. This manner is useful anticipating that S–Cd–Im CPs should have a very similar buffering behavior as Cd–Im CP which is an important property to consider for the photocatalysis application.

Raman spectra of imidazole, Cd–Im CP, and S–Cd–Im CPs
3.1.2 PXRD and SEM/EDX Analyses
Figure 3 shows PXRD patterns of Cd–Im CP and S–Cd–Im CPs. It is clear that Cd–Im CP and S–Cd–Im CPs have very similar PXRD patterns and triplet around 2θ 16.8 strongly gives surety. S–Cd–Im CPs are also of high crystalline nature as indicated from their sharp peaks. Principally, the crystalline structure of a semiconductor is advantageous for photocatalytic reactions due to the higher mobility of charge carriers [37]. The main peak of Cd–Im CP retains its position (17.1) and height in patterns of S–Cd–Im CPs indicating that the main matrix of CPs is intact after sulfur modification. The other smaller peaks retain their positions and this should be another indication of non-damage successful sulfur modification. Still, there are some significant alterations about 2θ = 30 and about 2θ = 44 to (dashed rectangles) which should be attributed to sulfur incorporation.

PXRD pattern of Cd–Im CP and S–Cd–Im CPs
According to Scherrer formula, the crystallite size of Cd–Im CP is 173.5 nm while crystallite sizes of 5S–Cd–Im CP and 10S–Cd–Im CPs are 288.9 and 247.8 nm respectively [38]. These sizes are of the same order and may indicate no great change in crystallite structure from Cd–Im CP to S–Cd–Im CPs. However, it is expected that some changes of lattice parameters (a,b and c) and unit cell volume had took place as a consequence of sulfur dopant inclusion [39]. The relatively increase of crystallite size of S–Cd–Im CPs should enhance light absorption [40], decrease the opportunity for the recombination of generated charges, and relieve the transfer of the light-generated carriers the surface of particle.
Figure 4 shows the SEM images of Cd–Im CP and S–Cd–Im CPs. Images show particles as top trimmed rhombic-bipyramid for all samples and this indicates the intact morphology. The particles’ sizes range from about 3 to about 5 μm on average which is appropriate for water treatment applications. From EDX data, sulfur to cadmium atom percentages are 0.65% and 0.81% for 5S–Cd–Im CP and 10S–Cd–Im CP respectively. Hence, it can be suggested that most sulphur modification effectively influenced particles superficially.

SEM images of a Cd–Im CP, b 5S–Cd–Im CP, and c 10S–Cd–Im CP
3.1.3 UV–Vis DRS Measurements and Bandgap Energies
UV–Vis DRS spectra of Cd–Im CP and S–Cd–Im CPs are shown in Fig. 5. Two bandgaps have been assigned for each of CPs. Eg values of Cd–Im CP are 4.55 and 3.4 eV whose corresponding wavelengths are 272 and 364 nm respectively. Hence, Cd–Im CP should be photocatalytically active under UV-lamp of λ 254 nm. Eg value 4.55 should reflect the π → π* transition of imidazole/imidazolate ligands within Cd–Im CP structure while Eg value 3.4 eV could reflect the metal to ligand charge transfer (MLCT) originates upon Cd/imidazolte formation [41].

Diffuse reflectance of a Cd–Im CP, b 5S–Cd–Im CP, and c 10S–Cd–Im CP
Upon superficial sulfur modification of the pristine Cd–Im CP forming S–Cd–Im CPs, the two bandgaps have significantly narrowed from 4.55 and 3.4 eV to 4.25 and 2.35 eV, respectively, for both the S–Cd–Im CPs. The first narrowing, 4.55 to 4.25 eV, could be attributed to the mixing of S 3p states with the VB of Cd–Im CP [37]. The second narrowing from 3.4 to 2.35 eV can be attributed to the suggested development Cd–S bonding. Cd–S bond could behave as the recognized CdS semiconductor (Eg(CdS) = 2.4 eV). These immerged CdS moieties can be seen as hetero-structure material that is in intimate coupling with original Cd–Im CP. Hence, the coupling of CdS (Eg 2.4 eV) and Cd–Im CP (4.55 and 3.4 eV) forms the CdS/Cd–Im CP composite. This coupling results in narrowing the original bandgap and could also reduce e−/h+ recombination due to intimate attachment of CdS with structure. It may act as a rapid separation site for the generated e–/h+ due to the difference in the energy level of their conduction bands and valence bands [35, 37, 42, 43]. Some similar results have been given in literature. Gascon et al.showed that the bandgap of MOF-5 can be modulated within the range of 3.3–4.0 eV by adding electron-donating substituents onto the organic linker [44]. Also, Espen Flage-Larsen et al.addressed the bandgap modification of the UiO-6x-R (x = 6, R = H, NH2, and NO2; x = 7, R = H; x = 8, R = H) structures. Upon substituting H with NH2 and NO2 on the linker, a significant reduction of the bandgap was observed by introducing NH2 and NO2, with different extents, in the structure [45].
On the whole, the new narrower bandgaps of S–Cd–Im CP, 2.35 and 4.25 eV, correspond to the wavelengths 291 and 527 nm respectively and indicate that S–Cd–Im CPs should be photoactive under both UV-light and Visible-light via an indirect mechanism.
3.1.4 TGA Analyses
TGA thermograms of Cd–Im CP and S–Cd–Im CPs are shown in Fig. 6. Cd–Im CP shows no significant mass loss up to 150 °C where all water content starts to escape (about: 6%), in the temperature range of 158 °C to 200 °C. Meanwhile, water loss for S–Cd–Im CPs starts at about 275 °C. Gradual, yet not smooth, decomposition starts at 200 °C for Cd–Im CP and at about 300 °C for S–Cd–Im CPs and ceased for all CPs at about 700 °C. Mass loss in these ranges should be due to of the decomposition of organic and hydroxyl ligands (about: 47%). Generally, these behaviors indicate that S-treatment strengths S–Cd–Im CPs matrix structures (at least the outer layers where sulfur is mostly presented). Also, the non-smooth decomposition reflects the different groups bonded to Cd in CPs structure.

The TGA curves for Cd–Im CP and S–Cd–Im CPs
3.1.5 Stability of Cd–Im CP in Water
According to mass-loss test, the hydro-stability of Cd–Im CP is considerable as the mass loss was almost insignificant. Hence, no hydrolysis or functionality release could be expected. In addition, the FTIR spectra of the pristine and in-water soaked samples, Fig. 7, are nearly similar which indicates that the soaked sample retains generally its chemical structure where almost all peaks preserve its position. In general, frameworks constructed by M2+ and imidazolate linkers are known to be hydro-stable [12].

FTIR spectra of the pristine and soaked Cd–Im CP
4 Photocatalytic Activity of Cd–Im CP and S–Cd–Im CPs
It should be declared the consideration that the observed measured degradation of a photocatalyzed experiment is in fact the degradation-resultant from both the photocatalysis and photolysis where the latter should proceed regardless a catalyst is present or not [46]. Further, at least one out of two mechanisms should be responsible for photocatalytic degradation, the direct one (D) which starts with self-sensitization and includes dye-adsorption, and the indirect one (In-D) which starts with the known catalyst excitation (eCB−/hVB+ formation).
Cd–Im CP has a buffering behavior and has an operating pH of 8.5 and D mechanism is affected by this pH condition [32]. Consequently, buffering behavior of Cd–Im CP should be also considered for mechanistic explanation and might be responsible for degradation similarity in the present work. Still, also the presence of a dye molecule within a reasonable distance from catalyst surface and with a suitable orientation cause the formation of transitional activated dye-complex which eventually suffers degradation [32, 47,48,49]. The confirmation is that degradation of different organic molecules occurs non-selectively, but depending on the closeness to the catalyst surface. This illustration can cause the degradation of dyes regardless of their ionic type [50].
4.1 Photocatalytic Degradation of MB and MO Under Visible-Light
Under Visible-light, only the D mechanism is in operation for Cd–Im CP while D and In-D mechanisms are operating for S–Cd–Im CPS due to bandgap modification by sulfur. Figure 8a shows the degradations of MB due to photolysis and photocatalysis under Visible-light by Cd–Im CP and S–Cd–Im CPs for the different initial pH 4, 7 and 10.

Photocatalytic and photolytic degradations of a MB and b MO at different initial pH levels under Visible-light
For initial pH 4, photolysis degradation and photocatalysis degradation by Cd–Im CP are similar and the conclusion is that photocatalysis does not play an effective role. Hence, D mechanism is not operating and this is due to the extremely strong repulsion of MB+ by the CP of the highly positive surface (pH changes from 4 to 8.5, ∆pH = 4.5), i.e. neither dye adsorption nor closeness to the surface is plausible to initiate this mechanism. Meanwhile, S–Cd–Im CPs show about threefold degradations higher than that of Cd–Im CP. This should be to great extent due to the bandgap modification that induces operating In-D mechanism initiating formation of the oxidizing˙O2– species and maybe to some extent, the lesser positivity of surface due to sulfur doping.
For initial pH 7, photocatalysis by Cd–Im CP is slightly higher than photolysis. This is due to the neutrality of the surface, where the D mechanism has the opportunity to operate. S–Cd–Im CPs show a further increase in degradation as both mechanisms, D and In-D, are in operation.
For initial pH 10, photolysis is oddly better than photocatalysis by Cd–Im CP. This is a strange phenomenon and means that the presence of Cd–Im CP catalyst inferiors photolysis. The buffering behavior of Cd–Im CP should be the cause of this phenomenon. During an early buffering event for initial pH 10, Cd–Im CP exceedingly consumes the supplied OH– which effectively reduces the production of the oxidizing radical ˙OH, considering In-D mechanism, required for efficient photolysis degradation. In the meantime, the D mechanism is prohibited to great extent because of the strong screening positive Na+-stern layer covering the Cd–Im CP surface. Nonetheless, S–Cd–Im CPs show significant better degradation than photolysis and photocatalysis by Cd–Im CP and two reasons should be behind this: (i) the effective involvement of In-D mechanism, where freshly originated oxidizing ˙O2– species are strongly repelled towards a bulk solution, and (ii) OH– consumption is lowered due to sulfur coordination which lowers OH–/surface interaction and hence photolysis reinstates to a certain extent [32]. Hence, degradation was enhanced by S–Cd–Im CPs to the extent that complete destroy of MB occurred within only about 2 h by 10S–Cd–Im CP.
Figure 8b shows the degradations of MO due to photolysis and photocatalysis under Visible-light by Cd–Im CP and S–Cd–Im CPs for different initial pH 4, 7 and 10. For initial pH 4, photocatalysis by Cd–Im CP takes place via the D mechanism and is higher than photolysis. This is due to the positive surface albeit facing some resistance from the negative Cl–-stern layer. S–Cd–Im CPs show a further increase of degradation due to parallel engagement of the two degrading mechanisms, D and In-D.
For initial pH 7, photocatalysis by Cd–Im CP is rather higher than photolysis due to the enhanced D mechanism because of the neutral surface with no ionic cloud resistance as no stern layer forms. Again, S–Cd–Im CPs show a further increase of degradation due to parallel engagement of the two degrading mechanisms.
For initial pH 10, photocatalysis by Cd–Im CP is higher than photolysis although the surface is negative. However in this case, negativity is not that strong against MO– (pH changes from 10 to 8.5, ∆pH = 1.5) as positivity against MB+ for initial pH 4 (∆pH = 4.5). Hence, the positive Na+-stern layer gives the chance for MO to surge/touch CP surface to initiate the D mechanism. And again, S–Cd–Im CPs show a further increase of degradation due to parallel engagement of the two mechanisms, D and In-D.
Yet, lasting is important to present results of the pure effect of photocatalysis, Dp%, under Visible-light, i.e. by eliminating the parallel photolysis effect, to elucidate CPs’ worth. Figure 9 presents these degradation results after 5 h. From the Fig. 9, S–Cd–Im CPs are appreciably more degrading than Cd–Im CP for all initial pH conditions and activity increases with initial pH decrease for the both dyes irrespective of their charges. Hence, sulfur doping is effective in enhancing degradation under Visible-light and the In-D mechanism is the more effective enrolling one [35]. Imperative is the increase of degradation by S–Cd–Im CPs with solution acidity, irrespective of the charge type of dyes. This should point at the importance of migration-strength of ˙O2– radicals toward the bulk solution to accomplish degradation, respective to the In-D mechanism, and this should be attributed to the nature of produced stern-layers. [32]. To evaluate degradation performance quantitatively, the highest apparent turnover frequencies, TOF, have been calculated for the best results given by 10 S–Cd–IM CP and these are as follows: 1.21 × 10−3 h−1 for MB at initial pH 10 and 1.03 × 10−3 h−1 for MO at initial pH 4.

Effective photocatalysis, % Dp, under Visible-light after 5 h (elimination of photolysis effect), A MB, and B MO
4.2 Photocatalytic Degradation of MB and MO Under UV-Light
Under UV-light, both D and In-D mechanisms are in operation for both Cd–Im CP and Cd–Im CPs. Figure 10a shows the degradations of MB due to photolysis and photocatalysis under UV-light by Cd–Im CP and S–Cd–Im CPs for different initial pH 4, 7 and 10.

Photocatalytic and photolytic degradations of a MB and b MO at different initial pH levels under UV-light
For the initial pH 4, surprisingly, all photocatalysis, either by Cd–Im CP or by S–Cd–Im CPs, show lower values than that of photolysis which means that presence of any CP depressed photolysis mechanism (This is in contrast to that of Visible-light, except for Cd–Im CP at initial pH 10 case). This can be correlated to the buffering of CPs and the catalyst excitation due to UV-light. For diluted solutions, solutes do not powerfully respond to radiation (excitation/ionization), however, on the other hand, water gives the following assortment of radiolytic products [51]:
Species like .OH, H2O2 and .O2– are oxidizing and should be considered. However, the most important oxidizing radical is the superoxide anion, .O2–. In addition to water as a source of .O2– and .OH, another source of .O2– is the In-D mechanism; knowingly .O2– and .OH should also be produced on catalyst surface \( \left( {{\text{O}}_{{{\text{2 dissolved}}}} + {\text{ e}}_{{{\text{CB}}}} ^{{-}} \to {^{{\mathbf{.}}} {\text{O}}_{{\text{2}}}}^{{-}} _{{{\text{ads}}}} \,{\text{and}}\,{\text{ H}}_{{\text{2}}} {\text{O }} + {\text{ h}}^{ + } ~~~ \to ~~~^{{\mathbf{.}}} {\text{OH}}} \right) \) [2]. One proposal for photolysis depression is based on catalyst buffering and water radiolysis. Buffering creates an exceedingly positive surface that sturdily attracts .O2– from the water source part and also confines .O2– from In-D source part dawdling their diffusion outwards to attack MB in bulk solution. Following that is the continuous exhaustion of .O2– and also H2O2 and .OH conversions into H2O, by the water radiolysis products (eaq–, 2H+) according to proposed following reactions [52]:
As the equation shows, exhaustion and conversion require an abundance of H+. In addition to the direct radiolysis of water as H+ source, the following catalyst-dependent reaction is another continuous source of H+ [53]:
Interestingly, as sulfur content increases depression weakens, i.e. photolysis recurs. It can be suggested that the rate of radicals’ quenching to H2O starts to restrain by extra production of .O2– on S–Cd–Im CPs as sulfur becomes an eCB– collecting/stabilizing site.
For initial pH 7, photocatalysis is clearly higher than photolysis and the effect slightly increases with the increase of sulfur content. This is because of the neutrality of CPs’ surfaces and the faded ionic cloud which gives .O2– the opportunity to migrate towards the bulk performing its oxidizing effect.
Actually, for initial pH 10, photolysis is already extremely high and photocatalysis are not that higher than photolysis. This should be due to some repelling by the positive Na+-stern layer, decelerating D mechanism.
Figure 10b shows the degradations of MO due to photolysis and photocatalysis under UV-light by Cd–Im CP and S–Cd–Im CPs for different initial pH 4, 7 and 10. From the figure, photocatalysis by Cd–Im CP and S–Cd–Im CPs are very similar and hence sulfur doping is not that effective for MO under UV-light radiation. Remarkably, degradation by photocatalysis by any of the applied CPs is higher than degradation by photolysis especially for initial pH 7 elucidating the role of buffering nature of CPs, their surface charge, and stern layer ion population. In other words, when the surface charge is neutral with a faded stern layer, photocatalysis goes easily via D mechanism. Also, these conditions allow readily migration of .O2– (formed on CP surface upon In-D mechanism) towards a bulk solution.
For initial pH 4 and compared to pH 7 case, although the surface is sought as highly positive and should strongly attract MO–, negative Cl−-stern layer seems to decelerate this attraction and hence degradation. However and herein, it is important to draw attention to the different behaviors of degrading MB and MO considering pH 4. As mentioned earlier, MB degradation by photolysis is higher than those by CPs and depression mechanism of photolysis was suggested via continuous exhaustion of the formed˙O2–. In contrast, MO degradation by CPs is effective than by photolysis. Hence, dye charge should play a central role in degradation process under this pH condition. Depression mechanism of photolysis should also present for MO case, however I seems that MO-negativity charge helps in giving MO molecules some nearness towards CPs surface giving some chance for degradation with some available oxidizing species.
For initial pH 10, the surface is highly negative and should repel MO–, however surface repel also the In-D generated .O2– towards the bulk solution, and in the meantime the formed positive Na+-stern layer gives the chance for some sneaky MO– migration towards CPs’ surfaces which eventually originates degradation.
The very obvious effect of photocatalysis over photolysis encourages the use of these CPs for neutral and alkaline solutions laden with anionic organic contaminants. Most importantly, S–Cd–Im CPs show relatively slight enhancement in degradation compared to Cd–Im CP.
Again, it is important to present results of the pure effect of photocatalysis under UV-light, i.e. photolysis elimination, to elucidate CPs’ worth. Figure 11 presents these degradation results after 5 h. From the Fig. 11, S–Cd–Im CPs are generally barely more degrading than Cd–Im CP for all initial pH conditions. Hence, sulfur doping is not that effective in enhancing degradation under UV-light.

Effective photocatalysis, Dp%, under UV-light after 5 h (elimination of photolysis effect), A MB, and B MO
5 Conclusions
The majority of CPs and MOFs photocatalysts, in their pristine structure, has a wide bandgap and high electron–hole recombination and consequently can absorb only UV-light. Therefore, it is necessary to develop some strategies to activate such CPs and MOFs to be able to efficiently harvest visible-light with suitable derived photocaltalysis. In this work, doping of a pristine CP by a non-metal is considered as a novel strategy, where Cd–Im CP was treated by sodium sulfide to embrace sulfur as a chemical dopant. From FTIR and Raman analysis, sulfur treatment of Cd–Im CP successfully produced chemically sulfur-doped samples, S–Cd–Im CPs. By doping, the pristine crystalline phase has not been observed to change, according to PXRD, which helps in preserving photocatalysis nature. However, importantly, doping by the non-metal sulfur did significantly alter pristine bandgaps from 4.55 and 3.4 eV to 4.25 and 2.35 eV for S–Cd–Im CPs and the latter allow active photocatalysis under both UV-light and Visible-light via the indirect mechanism. Although degradation under UV-light gives the highest values, S–Cd–Im CPs performance is similar to that of Cd–Im CP which means that sulfur doping does not significantly promote photocatalysis under this type of light. Meanwhile, S–Cd–Im CPs performance under Visible-light generally multiplies degradation compared to that of Cd–Im CP which means that sulfur doping significantly enhances photocatalysis under Visible-light. As for buffering materials (highly charged surface and highly ion-populated stern layer for initial pH 4 and 10), these CPs control photocatalytic degradation in different manners and should be considered carefully. Because of buffering, it is found that surface charge and stern-layer charge play important role. They are opposing in attracting/repelling ionic molecules towards CP-surface with surface charge being slightly more effective. The phenomenon of photolysis depression, for certain initial pH, is striking and should be considered for similar photocatalysts to study carefully and deeply. In fact, the performance of S–Cd–Im CPs under Visible-light is not that competitive with other hybrid photocatalysts. However this work highlights the non-metal doping as a promising strategy to enhance the photoresponse of the hybrid materials and the given mechanisms encourage for extensive researches for enhanced non-metal doped hybrid photocatalysts. Further studies should be carried out considering other types of non-metal dopants and central is the research for better understanding of the origin of photoresponse enhancement on molecular/structural basis.
Data Availability
Not applicable.
References
X. Zhang et al., Functionalized metal-organic frameworks for photocatalytic degradation of organic pollutants in environment. Chemosphere 242, 125144 (2020)
Q.-Q. Xiao et al., Two difunctional Co (II) coordination polymers for natural sunlight photocatalysis of methylene blue and selective fluorescence sensing of Cr (VI) ion in water media. J. Solid State Chem. 276, 331–338 (2019)
Y. Wu et al., Design and construction of diverse structures of coordination polymers: photocatalytic properties. J. Solid State Chem. 245, 213–218 (2017)
K.K. Bisht et al., Mixed ligand coordination polymers with flexible bis-imidazole linker and angular sulfonyldibenzoate: crystal structure, photoluminescence and photocatalytic activity. J. Solid State Chem. 213, 43–51 (2014)
S. Zinatloo-Ajabshir et al., Facile preparation of Nd2Zr2O7–ZrO2 nanocomposites as an effective photocatalyst via a new route. J. Energy Chem. 26(2), 315–323 (2017)
S. Mortazavi-Derazkola, S. Zinatloo-Ajabshir, M. Salavati-Niasari, New facile preparation of Ho2O3 nanostructured material with improved photocatalytic performance. J. Mater. Sci.: Mater. Electron. 28(2), 1914–1924 (2017)
M.S. Morassaei, S. Zinatloo-Ajabshir, M. Salavati-Niasari, Simple salt-assisted combustion synthesis of Nd2Sn2O7–SnO2 nanocomposites with different amino acids as fuel: an efficient photocatalyst for the degradation of methyl orange dye. J. Mater. Sci.: Mater. Electron. 27(11), 11698–11706 (2016)
Z. Salehi, S. Zinatloo-Ajabshir, M. Salavati-Niasari, Novel synthesis of Dy2Ce2O7 nanostructures via a facile combustion route. RSC Adv. 6(32), 26895–26901 (2016)
S. Mortazavi-Derazkola, S. Zinatloo-Ajabshir, M. Salavati-Niasari, Facile hydrothermal and novel preparation of nanostructured Ho2O3 for photodegradation of eriochrome black T dye as water pollutant. Adv. Powder Technol. 28(3), 747–754 (2017)
S. Zinatloo-Ajabshir, S. Mortazavi-Derazkola, M. Salavati-Niasari, Schiff-base hydrothermal synthesis and characterization of Nd2O3 nanostructures for effective photocatalytic degradation of eriochrome black T dye as water contaminant. J. Mater. Sci.: Mater. Electron. 28(23), 17849–17859 (2017)
F. Razi, S. Zinatloo-Ajabshir, M. Salavati-Niasari, Preparation, characterization and photocatalytic properties of Ag2ZnI4/AgI nanocomposites via a new simple hydrothermal approach. J. Mol. Liq. 225, 645–651 (2017)
Q. Wang et al., Recent advances in MOF-based photocatalysis: environmental remediation under visible light. Inorg. Chem. Front. 7(2), 300–339 (2020)
X. Quan et al., Polyethyleneimine (PEI) incorporated Cu-BTC composites: Extended applications in ultra-high efficient removal of congo red. J. Solid State Chem. 270, 231–241 (2019)
E. Whelan et al., Tuning photoactive metal–organic frameworks for luminescence and photocatalytic applications. Coord. Chem. Rev. 437, 213757 (2021)
C.R. McKeithan, L. Wojtas, R.W. Larsen, Guest to framework photoinduced electron transfer in a cobalt substituted RWLC-2 metal organic framework. Dalton Trans. 47(28), 9250–9256 (2018)
O. Abuzalat et al., Advances of the highly efficient and stable visible light active photocatalyst Zr (iv)–phthalate coordination polymer for the degradation of organic contaminants in water. Dalton Trans. 50(24), 8600–8611 (2021)
J.-S. Qin et al., Stable metal–organic frameworks as a host platform for catalysis and biomimetics. Chem. Commun. 54(34), 4231–4249 (2018)
Z.U. Zango et al., A critical review on metal-organic frameworks and their composites as advanced materials for adsorption and photocatalytic degradation of emerging organic pollutants from wastewater. Polymers 12(11), 2648 (2020)
Y.K. Hwang et al., Amine grafting on coordinatively unsaturated metal centers of MOFs: consequences for catalysis and metal encapsulation. Angew. Chem. 120(22), 4212–4216 (2008)
M. Alvaro et al., Semiconductor behavior of a metal-organic framework (MOF). Chem.: Eur. J 13(18), 5106–5112 (2007)
R. Liang et al., MIL-53 (Fe) as a highly efficient bifunctional photocatalyst for the simultaneous reduction of Cr (VI) and oxidation of dyes. J. Hazard. Mater. 287, 364–372 (2015)
G. Zahn et al., A water-born Zr-based porous coordination polymer: modulated synthesis of Zr-fumarate MOF. Microporous Mesoporous Mater. 203, 186–194 (2015)
R.M. Abdelhameed et al., Enhanced photocatalytic activity of MIL-125 by post-synthetic modification with CrIII and Ag nanoparticles. Chem.: Eur. J. 21(31), 11072–11081 (2015)
A. Wang et al., Titanium incorporated with UiO-66 (Zr)-type Metal-Organic Framework (MOF) for photocatalytic application. RSC Adv. 6(5), 3671–3679 (2016)
R. Li et al., Nickel-substituted zeolitic imidazolate frameworks for time-resolved alcohol sensing and photocatalysis under visible light. J. Mater. Chem. A 2(16), 5724–5729 (2014)
J.A. Botas et al., Effect of Zn/Co ratio in MOF-74 type materials containing exposed metal sites on their hydrogen adsorption behaviour and on their bandgap energy. Int. J. Hydrog. Energy 36(17), 10834–10844 (2011)
S. Chen et al., N-doped Cu-MOFs for efficient electrochemical determination of dopamine and sulfanilamide. J. Hazard. Mater. 390, 122157 (2020)
S.-I. Noro, T. Nakamura, Fluorine-functionalized metal–organic frameworks and porous coordination polymers. NPG Asia Mater. 9(9), e433–e433 (2017)
W. Yuan et al., Laminated hybrid junction of sulfur-doped TiO2 and a carbon substrate derived from Ti3C2 MXenes: toward highly visible Light-driven photocatalytic hydrogen evolution. Adv. Sci. 5(6), 1700870 (2018)
S.A. Ansari et al., Nitrogen-doped titanium dioxide (N-doped TiO 2) for visible light photocatalysis. New J. Chem. 40(4), 3000–3009 (2016)
P.S. Basavarajappa et al., Recent progress in metal-doped TiO2, non-metal doped/codoped TiO2 and TiO2 nanostructured hybrids for enhanced photocatalysis. Int. J. Hydrog. Energy 45(13), 7764–7778 (2020)
M. Mohsen et al., A cadmium-imidazole coordination polymer as solid state buffering material: synthesis, characterization and its use for photocatalytic degradation of ionic dyes. J. Solid State Chem. 289, 121493 (2020)
A. Baraka et al., A new cationic silver (I)/melamine coordination polymer, [Ag2 (melamine)] n2n+: synthesis, characterization and potential use for aqueous contaminant anion exchange. J. Solid State Chem. 274, 168–175 (2019)
X.-J. Wei et al., Construction of a series of Zn (II) and Cd (II) coordination polymers bearing 1, 3-bis (benzimidazol-1-yl)-2-propanol ligands: syntheses, crystal structures, and sensing properties. J. Solid State Chem. 284, 121218 (2020)
C. Yu et al., Sol–gel derived S, I-codoped mesoporous TiO2 photocatalyst with high visible-light photocatalytic activity. J. Phys. Chem. Solids 71(9), 1337–1343 (2010)
T. Slager, C. Amberg, Infrared investigation of H2S adsorption and decomposition on alumina and on alumina supported molybdenum sulfide. Can. J. Chem. 50(21), 3416–3423 (1972)
R. Marschall, L. Wang, Non-metal doping of transition metal oxides for visible-light photocatalysis. Catal. Today 225, 111–135 (2014)
P. Scherrer, Bestimmung der inneren Struktur und der Größe von Kolloidteilchen mittels Röntgenstrahlen, in Kolloidchemie Ein Lehrbuch. (Springer, Berlin, 1912), pp. 387–409
S. Dastagiri et al., Induced dielectric behavior in high dense AlxLa1-xTiO3 (x= 0.2–0.8) nanospheres. J. Mater. Sci.: Mater. Electron. 30(22), 20253–20264 (2019)
A.B.D. Nandiyanto, R. Zaen, R. Oktiani, Correlation between crystallite size and photocatalytic performance of micrometer-sized monoclinic WO3 particles. Arab. J. Chem. 13(1), 1283–1296 (2020)
J. Cepeda, A. Rodríguez-Diéguez, Tuning the luminescence performance of metal–organic frameworks based on d 10 metal ions: from an inherent versatile behaviour to their response to external stimuli. CrystEngComm 18(44), 8556–8573 (2016)
F. Al Marzouqi, Y. Kim, R. Selvaraj, Shifting of the band edge and investigation of charge carrier pathways in the CdS/gC3N4 heterostructure for enhanced photocatalytic degradation of levofloxacin. New J. Chem. 43(25), 9784–9792 (2019)
A. Kudo, Z-scheme photocatalyst systems for water splitting under visible light irradiation. MRS Bull. 36(1), 32 (2011)
J. Gascon et al., Isoreticular MOFs as efficient photocatalysts with tunable bandgap: an operando FTIR study of the photoinduced oxidation of propylene. Chemsuschem 1(12), 981–983 (2008)
E. Flage-Larsen et al., Bandgap modulations in UiO metal–organic frameworks. J. Phys. Chem. C 117(40), 20610–20616 (2013)
T.-W. Tzeng et al., Photolysis and photocatalytic decomposition of sulfamethazine antibiotics in an aqueous solution with TiO2. RSC Adv. 6(73), 69301–69310 (2016)
Z.T. Yu et al., Water-insoluble Ag–U–organic assemblies with photocatalytic activity. Chem.: A Eur. J. 11(9), 2642–2650 (2005)
A.K. Paul, G. Madras, S. Natarajan, Adsorption–desorption and photocatalytic properties of inorganic–organic hybrid cadmium thiosulfate compounds. Phys. Chem. Chem. Phys. 11(47), 11285–11296 (2009)
Q. Shang et al., A novel nitrogen heterocyclic ligand-based MOF: synthesis, characterization and photocatalytic properties. New J. Chem. 43(42), 16595–16603 (2019)
A. Ajmal et al., Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: a comparative overview. RSC Adv. 4(70), 37003–37026 (2014)
S. Le Caër, Water radiolysis: influence of oxide surfaces on H2 production under ionizing radiation. Water 3(1), 235–253 (2011)
P. Sutkowy, A. Woźniak, C. Mila-Kierzenkowska, Positive effect of generation of reactive oxygen species on the human organism. Medical and Biological Sciences 27(2), 13–18 (2013)
M. Tahir et al., Selective photocatalytic reduction of CO2 by H2O/H2 to CH4 and CH3OH over Cu-promoted In2O3/TiO2 nanocatalyst. Appl. Surf. Sci. 389, 46–55 (2016)
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Consent for Publication
All authors have agreed with the content, and all have given explicit consent to publish.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohsen, M., Tantawy, H., Naeem, I. et al. Activation of Cadmium–Imidazole Buffering Coordination Polymer by Sulfur-Doping for the Enhancement of Photocatalytic Degradation of Cationic and Anionic Dyes Under Visible Light. J Inorg Organomet Polym 32, 2961–2974 (2022). https://doi.org/10.1007/s10904-022-02324-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-022-02324-x