
Abstract
Purpose
Bacillus Calmette–Guerin (BCG) is a live attenuated vaccine with the potential of causing severe iatrogenic complications in patients with primary immunodeficiency diseases (PID) before and after hematopoietic stem cell transplantation (HSCT). We aim to investigate risk factors of post-HSCT BCG-related complications in PID patients.
Methods
A retrospective analysis of pediatric PID patients who had received the BCG vaccine and underwent HSCT at Hadassah-Hebrew University Medical Center, between 2007 and 2019.
Results
We found 15/36 (41.67%) patients who developed post-HSCT BCG-related complications. The most significant risk factor for developing BCG-related complications was T cell deficiency (47.6% of the non-complicated vs 83.3% of the BCGitis and 100% of the BCGosis groups had T cell lymphopenia, p = 0.013). None of the chronic granulomatous patients developed BCG-related manifestation post-transplant. Among T cell–deficient patients, lower NK (127 vs 698 cells/μl, p = 0.04) cell counts and NK-SCID were risk factors for ongoing post-HSCT BCGosis, as was pretransplant disseminated BCGosis (33.3% of patients with BCGosis vs none of the non-BCGosis patients, p = 0.04). Immune reconstitution inflammatory syndrome (IRIS) was observed in 3/5 patients with Omenn syndrome. Prophylactic antimycobacterial treatment was not proven effective.
Conclusion
BCG vaccination can cause significant morbidity and mortality in the post-transplant T cell–deficient patient, especially in the presence of pre-transplant disease. Taking a detailed medical history prior to administering, the BCG vaccine is crucial for prevention of this complication.
Similar content being viewed by others
Introduction
Mycobacterium tuberculosis (MTB) is a significant worldwide health threat [1]. Bacillus Calmette–Guerin (BCG) vaccine has been used widely since 1921 to prevent tuberculosis (TB) [1, 2], and a large proportion of the world’s population is vaccinated with BCG soon after birth. Nevertheless, the estimated prevalence of BCG side effects is 1:2500 and 1:100,000 for localized and disseminated complications, respectively [3]. BCG-related complications can be subdivided into local (vaccine area) or regional (adjacent lymph-node) vaccine-related complications referred to as BCGitis, versus distant (affecting one site) or disseminated (affecting > 1 site and/or blood) BCG infection, referred to as BCGosis [4, 5]. Either pattern can be a manifestation of an ongoing infection or can represent an immune reconstitution inflammatory syndrome (IRIS).
Since BCG is a live attenuated vaccine, complication rates are significantly increased in immunodeficient individuals [3, 6,7,8,9,10,11], especially so in patients with T cell defects, mendelian susceptibility to mycobacterial disease (MSMD), or chronic granulomatous disease (CGD) [5,6,7, 11], reflecting the main mechanisms of defense against mycobacteria. For T cell defects, this susceptibly is related to the inability to produce an effective Th1 response [3]. MSMD patients suffer from defects in the interleukin (IL)-12/IL-23/interferon (IFN) γ circuit [12] and CGD, caused by defects in the NADPH oxidase complex, adversely affect the macrophage’s and neutrophil’s ability to kill phagocytosed bacilli [6, 13]. Severe combined immunodeficiency (SCID) is the most common diagnosis among BCG-complicated primary immunodeficiency (PID) patients [3, 6] (42–51% of BCG-vaccinated SCID patients), with the highest mortality rate (41.2%) [6]. Lower T cell counts, younger age at vaccination [3], and natural killer (NK)-SCID [6] are risk factors for BCG-related complications.
Little is known about the prevalence and risks for BCG-related complications in the post-transplant setting. In this study, we summarize our experience with BCG vaccine-related complications in PID patients in the post-hematopoietic stem cell transplantation (HSCT) period.
Materials and Methods
This study was conducted at Hadassah Hebrew University Medical Center in Jerusalem. We retrospectively collected data from pediatric patients with inborn phagocytic or T cell defects who underwent HSCT between January 2007 and December 2019, after having received the BCG vaccine. Patients were divided into combined (T cell mediated) and phagocytic defects based on the international union of immunological societies (IUIS) classification of PIDs [14]. SCID was defined in T cell–deficient patients, who presented in the first year of life and had CD3 counts < 500 cells/μl [15]. PT9 without available pre-HSCT CD3 count, but with IL2RG mutation and a classical SCID phenotype, was also considered a SCID patient.
All patients had received their vaccines within the first week of life, as per the Russian and Palestinian schedules. A total of 32 of 36 patients had been vaccinated in the Palestinian Authority with the BCG Danish-1331 strain. Four patients (P9, P21, P32, and P35) had received the Moscow-368 strain in Russia. Prophylactic antimycobacterial treatment was given as per our protocol, which varied over time. Since 2013, patients with severe T cell deficiency and a history of BCG vaccine were given triple therapy (isoniazid, rifampin, and ethambutol), while rifampin was substituted during HSCT with ciprofloxacin to avoid drug interactions. Treatment of symptomatic BCG complications was tailored per patient. In general, antimycobacterial treatment was intensified, and in cases of lack of improvement under such a regimen, along with signs of hyperinflammation (fever, increase in inflammatory markers) steroid treatment was added.
We recorded baseline patient characteristics, timing, and nature of BCG complications, prophylactic and treatment regimens, immunological work up, transplant characteristics, and outcome. Graft-versus-host disease (GVHD) grading was based on the Glucksberg grading [16]. Genetic diagnosis was made via whole exome sequencing (WES) in 30 patients and confirmed by Sanger sequencing as described elsewhere [17]. For patients who were of the same kindred, diagnosis was done via WES in the index case and confirmed in family members by Sanger sequencing. Patients 25–26 were diagnosed via a CGD genetic panel. BCG-related complications were diagnosed based on clinical assessment of the treating physicians, supplemented with microbiology and pathology results whenever available. Complications were categorized as “infectious” when pre-transplant symptomatic disease further disseminated in the context of increased immune suppression, or disease manifestation emerged in the context of reduced immune function, or “inflammatory” when symptoms appeared for the first time shortly (within 45 days) after engraftment. In cases without symptomatic pre-transplant BCGosis, in which symptoms appeared prior to engraftment, it was impossible to discern worsening infection in the context of immune suppression from inflammatory reaction of donor T-cells, and therefore, we deemed these cases as “undetermined.” BCGitis was defined as local and/or the regional lymph node BCG infection, while BCGosis was defined as distant/disseminated disease [4].
Statistical analysis was conducted with the IBM_SPSS Statistics 27.0 version (IBM Inc., Armonk, NY). Cumulative survival curve was calculated using XLSTAT 2020.3.1 version. Fisher exact test was used for categorical variables. Mann-Withney U test and the Kruskal-Wallis H tests were used for 2 and 3 groups of independent numerical variables, respectively. Kaplan-Meier method was used for survival analysis. All statistical analyses conducted a 2-tailed test with a confidence level of 95%.

Cumulative Incidence for post-HSCT BCGosis. The cumulative risk of developing post-HSCT BCGosis in the first 500 days post-transplant. Cumulative incidence is 75.0% and 22.7% for patients with or without pre-transplant disease, respectively (p = 0.004)
This study was approved by Hadassah’s Institutional Review Board.
Results
Patient and transplant characteristics
Thirty-six pediatric patients who underwent HSCT for phagocytic and T cell defects in Hadassah medical center between 2007 and 2019, and had received the BCG vaccine prior to transplant, were included in this study (Table 1). Fifteen (41.7%) developed post-transplant BCG-related complications: 6 patients (40%) had BCGitis, while 9 patients (60%) had BCGosis. Twenty-four patients were transplanted for T cell defects, while 12 had phagocytic defects. Genetic diagnoses are detailed in Tables 2 and 3. Baseline diagnoses significantly differed between complication groups: while only 47.6% of the BCG-uncomplicated patients had a T cell deficiency, 83.3% of the BCGitis and all BCGosis patients had a T cell defect (Table 1). This difference in baseline immune defect accounts for non-complicated patients’ older age at the time of transplant compared with patients with BCGitis or BCGosis (49.2 versus 23.3 and 9.9 months old, respectively, p = 0.002), as well as the increased use of prophylactic antimycobacterial treatment in this group, considering our institutional protocol (66.7% in the BCGosis group vs 16.7% and 23.8% in the BCGitis and non-complication groups, respectively, p = 0.05). Pre-transplantation BCGosis was a predictor for post-transplant disseminated disease (75% and 22.7% cumulative incidence of post-transplant BCGosis among patients with and without pre-transplant disease, respectively, p = 0.004 Fig. 1).
Transplant variables are detailed in Tables 2 and 3. Donor, graft source and dose, conditioning regimen, and GVHD prophylaxis did not differ between groups. All transplants in this cohort were first transplants, and none of the patients required a second procedure.
Considering their increased rate of BCG-related complications, we analyzed T cell–deficient patients separately (Table 4). On a pooled analysis of BCGosis patients compared with non-complicated and BCGitis patients, those with BCGosis were younger at the time of transplant (mean age 9.9 vs 34.2 months, p = 0.045) and were more likely to have suffered from BCGosis prior to transplantation (33.3% of the BCGosis group vs none of the non-BCGosis group, p = 0.04). NK-cell counts prior to transplantation in BCGosis patients were significantly lower compared with the non-BCGosis group (243 vs 698 cells/μl, p = 0.03).
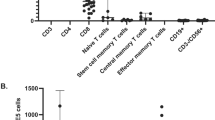
In order to delineate other risk factors, T cell–deficient patients without a pre-HSCT history of BCG infection were analyzed separately: those who developed BCGosis had lower pre-HSCT CD3 (32 vs 2425 cells/μl, p = 0.02) and NK-cell counts (127 vs 698 cells/μl, p = 0.04), but also a shorter diagnosis to transplant interval (mean time 2.2 vs 10.7 months, p = 0.02) (Fig. 2).

Pre-HSCT immune variables in T cell–deficient patients with post-HSCT BCGosis. Baseline immune variables of T cell–deficient patients without a prior history of pre-HSCT BCGosis. Patients who developed post-HSCT BCGosis are compared with those who did not develop this complication: a CD3+ counts were scaled on a logarithmic scale. b NK cell counts were scaled on a linear scale
Among eight asymptomatic T cell–deficient patients receiving antimycobacterial prophylaxis prior to transplantation, 50% developed post-transplant BCGosis versus two-thirteenths (15.4%) of untreated patients (p = 0.15). Prophylactic regimens did not differ between patients who developed BCGosis and those who did not (mean number of medications 2.75 vs 2.25, p = 0.12, mean duration of prophylaxis 0.59 vs. 0.75 months, p = 0.88). However, patients who developed BCGosis despite prophylaxis had JAK3 (n = 2), RAG1, and PNP deficiencies, whereas T cell–deficient, uncomplicated patients had the IL2RG c.664C > T variant (n = 3) and RAG1, suggesting a milder disease phenotype. On the other hand, both T cell–deficient patients receiving antimycobacterial treatment for pre-transplant symptomatic BCGosis experienced further deterioration of their disease despite appropriate treatment with 3–4 medications for a period of 1–4 months. Both patients subsequently died of disseminated disease 7–17 months post-transplant.
Younger age at transplantation and a shorter diagnosis to transplant interval, observed in T cell–deficient patients who developed post-HSCT BCGosis, are potential markers for a more severe baseline condition. We therefore analyzed the patients with SCID separately (n = 8, 5 (55.6%) of the BCGosis group and 3 (20%) of the non-BCGosis group, p = 0.099; Table 4). In this cohort, there was no difference in patient’s age at transplant or in diagnosis to transplant interval (mean age 10.8 vs 9.3, p = 0.55, mean time from diagnosis to treatment 2.2 and 1.7 months, p = 0.34, in the BCGosis and non-BCGosis groups, respectively). However, pre-HSCT NK-cell count remained significantly lower in the BCGosis group compared with the non-BCGosis group (47 vs 597 cells/μl, p = 0.03). Patients with SCID who developed BCGosis had mutations in JAK3 (n = 2), PNP, IL7R, and IL2RG, whereas those who did not develop this complication had mutations in RAG1 (n = 2) and LCK.
Among patients with phagocytic defects, only PT8 with VPS45 experienced mild BCGitis (Table 2). PT35 with CYBB mutation and pre-HSCT BCGosis was treated with triple therapy prior and during transplant and had not suffered reactivation of BCG. He is currently 12 months post-HSCT with full donor chimerism (Table 3).
Pathogenesis of BCG-Related Complications
Four patients presented with infectious complications: PT3, PT4, and PT10 had severe BCGosis prior to transplantation, and their infection disseminated further during transplant-related immunosuppression. Both PT3 and PT4 had viable bacilli in pathology or microbiological specimens (Table 2). While PT4’s duodenal biopsy revealed chronic mild inflammation without granulomas, PT3’s initial biopsies revealed granulomas which were absent in subsequent biopsies, suggesting a declining immune function [18]. In another patient (PT14), deteriorating immune function following a decline in donor chimerism had uncovered a BCG infection, as evident by positive gastric aspirate cultures and granulomas with acid-fast bacteria (AFB) on lymph-node (LN) biopsy.
Eight patients presented with inflammatory complications (Table 2). These were characterized by initial local/regional disease on average day + 34.4 (range 19–60 days) post-HSCT, followed by further dissemination in 2 patients. Another 3 patients (PT5, PT6, PT9) without pre-transplant disease had developed symptoms prior to engraftment; thus, it was impossible to differentiate donor cell allo-reactivity from unmasked previously asymptomatic infection in the context of immune suppression in these cases.
Five patients demonstrated a switch from positive to sterile cultures following antimycobacterial treatment: while 4 of these patients (PT1, PT2, PT9, and PT15) had evidence of granulomatous inflammation, suggestive of IRIS [18], PT5 exhibited a switch from a mononuclear infiltrate with positive Ziehl-Neelsen (ZN) stain on an early skin specimen, into a foamy histiocytic infiltrate with multinucleated giant cells on a subsequent specimen. These changes are suggestive of a transition from active infection to IRIS following engraftment [18]. Of 12 patients receiving DLI, only one (PT15) suffered further worsening of her skin BCGosis 34 days following DLI.
Patient Outcomes
Mean follow-up was 32.6 months (range 0–105 months) and did not differ significantly between groups (30.9, 53.8, and 22.6 months for non-complicated, BCGitis and BCGosis groups, respectively, p = 0.12). Patients with BCGosis had significantly lower donor chimerism on day + 100 compared with non-BCGosis patients (53.4 vs 93.4% donor cells, p = 0.01) and at last follow-up (47.4 vs 86.6% p = 0.02). This difference remained as a mere trend in T cell–deficient patients (donor chimerism on day + 100: 53.4 vs 87.4%, p = 0.09, and donor chimerism at last follow-up: 47.4 vs 78.2%, p = 0.097, in BCGosis compared with non-BCGosis patients), even after excluding patients without pre-transplant disease (donor chimerism on day + 100: 51.0 vs 87.4%, p = 0.09, and chimerism at last follow-up: 45.3 vs 78.2%, p = 0.08). T cell patients with BCGosis also had lower lymphocyte counts on day + 100 (2275 vs 4223 cells/μl, p = 0.02). Other outcome measurements did not differ between groups (Table 4).
Overall survival (OS) in our cohort was 83.3% (30/36 patients) and was comparable between T cell–deficient and phagocytic defect patients (79.2% and 91.7%, p = 0.41). In T cell–deficient patients, OS did not significantly differ among complication groups (OS = 80.0%, 100% and 66.7% in the non-complicated, BCGitis and BCGosis respectively, p = 0.39; Fig. 3). However, none of the T cell–deficient patients with a history of pre-HSCT BCGosis survived transplant, whereas survival rate without such history was 90.5% (Fig. 4; p < 0.001).

Survival in T cell deficiency patients with BCG-related complications. A Kaplan-Meier survival curve of BCG-complication groups in T cell–deficient patients demonstrates a similar survival: Overall survival (OS) = 80.0%, 100.0%, and 64.8% in patients with no BCG-related complications, BCGitis and BCGosis, respectively, p = 0.393

Survival in T cell deficiency patients with pre-HSCT BCGosis. A Kaplan-Meier survival curve of T cell–deficient patients demonstrates a significantly reduced survival in patients with a pre-transplant history of BCGosis compared with those lacking such history. Overall survival (OS) = 0% vs 90.5%, p < 0.001
All three patients with T cell defects who suffered of pre-transplant BCGosis died in the post-transplant period (Table 2): PT3 with inhibitor of nuclear kappa b kinase subunit beta (IKBKB) mutation was given a prolonged quadruple antimicrobial regimen for pre-transplant BCGosis. Following an MRD transplant, he suffered secondary graft failure and died of disseminated BCGosis and septic shock 17 months post-HSCT, despite continued anti-mycobacterial treatment. PT4 with IL7Rα deficiency was treated for 1 month prior to transplant for BCGosis. Following her MSD transplant she failed to reconstitute her T cells and responded poorly to antimicrobials. She died 7 months post-HSCT of multiple bowel perforations related to her BCG infection. PT10 with unidentified SCID presented with multiple bacterial infections. Upon admission, he was noted to have BCG lymphadenitis and hepatitis and an urgent mismatched unrelated (MMUD) cord blood (CB) was performed for lack of a better donor. Unfortunately, the patient failed to engraft and despite full antimycobacterial treatment succumbed to BCGosis as well as enteroviral infection. Three patients (PT27, PT31, and PT36) in the non-BCGosis group died in the early post-transplant period of veno-occlusive disease (VOD), sepsis, and non-engraftment (Table 3).
Discussion
In this retrospective observational study, we compare 15 PID patients with BCG vaccine-related post-HSCT complications to 21 BCG-vaccinated patients without such complications. Patients with T cell deficiencies were at an increased risk of developing BCG-related complications compared with phagocytic defect patients. Indeed, post-HSCT, BCG-related complications have been reported in T cell deficiencies [3, 18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] but have not so far been described in phagocytic defects. Despite the well-known association between CGD and BCG [6, 7, 13, 36], none of our 7 BCG-vaccinated CGD patients, including a patient with pre-transplant BCGosis, developed post-HSCT complications. These findings, in agreement with previous reports [13, 37], point to the central role of T cells in control of chronic mycobacterial infection and in mycobacterial-related IRIS [38].
History of pre-transplant disease is a major risk factor for post-HSCT BCGosis (Fig. 1; Table 4) as well as mortality (Fig. 4). In a review of the literature, 16/37 (43.2%) SCID patients with pre-transplant BCGosis died in the post-transplant period [18, 22,23,24, 27, 29, 31, 33, 39]. Increased mortality of pre-HSCT symptomatic patients was not found to be affected by antimycobacterial treatment [3]. Post-transplant treatment with anti-IL6 and Abatacept for prevention of inflammatory complications was recently reported in a small cohort [27] as both effective and safe. These results warrant further validation in larger, prospective studies.
Among T cell–deficient patients who had no pre-transplant BCGosis, lower baseline CD3 counts were found to be a risk factor for post-transplant BCGosis (Fig. 2). Poor T cell function was previously described as a risk factor for both BCG infection [3, 27] and future IRIS [40]. Lower NK-cell counts were found both in the T cell deficiency cohort, and in SCID patients specifically, and indeed, half of the patients in our SCID cohort who developed BCGosis had NK-SCID. This is compatible with a recent report [6], which identified a 2-fold higher number of NK negative SCID patients compared with NK positive SCID patients among the patients with BCG-related complications. NK cells have a role in control of mycobacterial infection, through the secretion of IFN-γ and TNFα as well as via direct contact [41, 42].
Various mechanisms have been ascribed to post-transplant BCG-related complications [6]. These can generally be divided into worsening of infection in the context of intensified immune suppression, versus increased inflammation in the context of an improved immune function. IRIS, defined as the restoration of an immune response against pathogen-specific antigens resulting in immunopathology, requires a functional immune system as a pre-requisite for diagnosis [43]. Features in our cohort consistent with IRIS included close association with engraftment and granuloma formation [18, 44], regardless of bacilli vitality. Features consistent with an infectious-driven process included active pre-transplant infection, poor immune reconstitution, and viable bacilli [18]. Nevertheless, distinguishing a predominant infectious process from an inflammatory reaction is challenging [45], since both mechanisms co-exist in the same patient during immune reconstitution. Gantzer et al. [18] demonstrated a switch from a diffuse histiocytic granuloma-negative infiltration with positive AFB to a CD3-rich, AF negative, granulomatous inflammation, as immunity was restored. Biopsy results can be helpful in distinguishing IRIS-related lesions from infectious worsening.
Three of five patients in our cohort who presented with Omenn syndrome developed IRIS. While there is not enough data to establish Omenn syndrome as a risk factor for post-HSCT BCG-related complications, it is possible that the switch from a Th2 milieu in Omenn syndrome [46], known to inhibit mycobacterial immune response [38], to a Th1 milieu in the post-HSCT period increased the risk of developing IRIS. Certainly, a shift from a Th2 to Th1/Th17 predominance is known to occur in multiple settings associated with IRIS [44, 45, 47].
There is no consensus regarding prophylactic treatment in PID patients [3]. In a previous retrospective analysis [3], only 3% of SCID patients receiving secondary prophylaxis developed BCG-related complications. However, prophylactic treatment did not improve OS. In our hands, post-exposure prophylaxis did not mitigate the risk for developing BCGosis. Nevertheless, as prospective controlled studies are lacking, we continue recommending a triple prophylactic regimen for profoundly T cell–deficient patients who have received the BCG vaccine. Another point to consider in prospective studies is the use of prophylaxis against inflammatory complications, as suggested in a recent report [27].
There are currently no guidelines for treatment of post-HSCT BCG disease, and diverse regimens have been used [3, 18,19,20,21, 24,25,26,27,28, 31, 33, 35, 39, 48, 49]. Treatment modalities other than anti-microbials include steroids [27, 29, 44, 47, 48], surgical treatment, [19, 20, 23, 27, 48] IFNγ, [18] and anti-IL1 and anti-IL-6R antagonists [27]. Our treatment protocol includes a triple regimen until clinical and radiological resolution of disease, as well as immune reconstitution. A recent report [27] eases safety concerns regarding anti-inflammatory treatment for IRIS on immune reconstitution. However, further studies are needed to better define the efficacy, safety, and need of such treatments in the post-transplant settings.
Finally, the WHO recommends avoiding BCG vaccination for immunodeficient infants [50]. These guidelines, however justified, are difficult to implement, due to immunization timing. Furthermore, neonatal screening does not encompass the wide range of BCG-associated PIDs and is currently not available in the countries with the highest burden of mycobacterial disease. Recommendations to postpone vaccination in at risk infants [50,51,52] must be weighed against the global health impact of BCG vaccination. Recently, the COVID-19 epidemic has stimulated new interest in the BCG vaccine for its possible non-specific protective effects through trained innate immunity, though evidence of efficacy of BCG vaccination against COVID-19 is lacking [53, 54]. Thus, while vaccine delay across all populations is neither practical nor advisable, we recommend that the possibility of PID should be considered prior to vaccination, especially in areas with a high prevalence of consanguinity.
Conclusion
BCG vaccination causes significant morbidity and mortality in PID patients, especially during post-transplantation immune reconstitution. Disseminated mycobacterial infection after HSCT is observed mainly in profound T cell deficiencies despite appropriate antibiotic treatment, with increased risk in NK-SCID. Pre-transplant infection is a significant negative prognostic factor. Taking a detailed medical and family history prior to administering the BCG vaccine is crucial and possibly lifesaving. Finally, there is a need for consensus guidelines for the prophylaxis and treatment of BCG disease in PID patients both prior to and following HSCT.
References
WHO. Global tuberculosis report 2019. WHO; 2020
Liu J, Tran V, Leung AS, Alexander DC, Zhu B. BCG vaccines: Their mechanisms of attenuation and impact on safety and protective efficacy. Hum Vaccin. 2009;5:70–8.
Marciano BE, Huang CY, Joshi G, Rezaei N, Carvalho BC, Allwood Z, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol. 2014;133(4):1134–41 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24679470.
Hesseling AC, Rabie H, Marais BJ, Manders M, Lips M, Schaaf HS, et al. Bacille Calmette-Guerin vaccine–induced disease in HIV-infected and HIV-uninfected children. Clin Infect Dis. 2006;42(4):548–58.
Ying W, Sun J, Liu D, Hui X, Yu Y, Wang J, et al. Clinical characteristics and immunogenetics of BCGosis/BCGitis in Chinese children: a 6 year follow-up study. PLoS One. 2014;9(4):e94485 [Internet]. [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24722620.
Fekrvand S, Yazdani R, Olbrich P, Gennery A, Rosenzweig SD, Condino-Neto A, et al. Primary Immunodeficiency Diseases and Bacillus Calmette-Guérin (BCG)-vaccine–derived complications: a systematic review. J Allergy Clin Immunol Pract. 2020;8(4):1371–86 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32006723.
Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N. Bacillus Calmette-Guérin (BCG) complications associated with primary immunodeficiency diseases. J Infect. 2012;64:543–54.
Al-Hammadi S, Alsuwaidi AR, Alshamsi ET, Ghatasheh GA, Souid AK. Disseminated Bacillus Calmette-Guérin (BCG) infections in infants with immunodeficiency. BMC Res Notes. 2017;10(1):177. https://doi.org/10.1186/s13104-017-2499-7.
Sadeghi-Shanbestari M, Ansarin K, Maljaei SH, Rafeey M, Pezeshki Z, Kousha A, et al. Immunologic aspects of patients with disseminated bacille Calmette-Guerin disease in north-west of Iran. Ital J Pediatr. 2009;35(42):42 [Internet]. [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20030825.
Lee W-I, Huang J-L, Yeh K-W, Jaing T-H, Lin T-Y, Huang Y-C, et al. Immune defects in active mycobacterial diseases in patients with primary immunodeficiency diseases (PIDs). J Formos Med Assoc. 2011;110(12):750–8 [Internet]. [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22248828.
Pöyhönen L, Bustamante J, Casanova JL, Jouanguy E, Zhang Q. Life-threatening infections due to live-attenuated vaccines: early manifestations of inborn errors of immunity [Internet]. J Clin Immunol. 2019;39:376–90 Springer New York LLC. [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31123910.
De Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine. 2010;89:381–402.
Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138(1):241–248.e3 [Internet]. [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26936803.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128.
Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130(1):177–83.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from hl-a-matched sibling donors1. Transplantation. 1974;18(4):295–304.
Stepensky P, Keller B, Shamriz O, NaserEddin A, Rumman N, Weintraub M, et al. Deep intronic mis-splicing mutation in JAK3 gene underlies T-B+NK- severe combined immunodeficiency phenotype. Clin Immunol. 2016;163:91–5.
Gantzer A, Neven B, Picard C, Brousse N, Lortholary O, Fischer A, et al. Severe cutaneous bacillus Calmette-Guérin infection in immunocompromised children: the relevance of skin biopsy. J Cutan Pathol. 2013;40(1):30–7 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23157280.
Erbey F, Dur AH, Atay D, Akçay A, Tecimer T, Öztürk G. Successful treatment of BCG-related immune reconstitution inflammatory syndrome following ex vivo T-cell-depleted haploidentical hematopoietic stem cell transplantation: a case report. Pediatr Transplant. 2019;23(5):e13464 [Internet]. [cited 2020 May 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31081274.
Abu-Arja RF, Gonzalez BE, Jacobs MR, Cabral L, Egler R, Auletta J, et al. Primary immunodeficiency diseases and Bacillus Calmette-Guérin (BCG)-vaccine–derived complications: a systematic review. J Allergy Clin Immunol Pract. 2020;8(4):1371–86 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32006723.
Aytekin C, Yuksek M, Dogu F, Yagmurlu A, Yildiran A, Fitoz S, Kologlu M, Babacan E, Hershfield MS, Ikinciogullari A. An unconditioned bone marrow transplantation in a child with purine nucleoside phosphorylase deficiency and its unique complication. Pediatr Transplant. 2008;12(4):479–82. https://doi.org/10.1111/j.1399-3046.2007.00890.x.
Amayiri N, Al-Zaben A, Ghatasheh L, Frangoul H, Hussein AA. Hematopoietic stem cell transplantation for children with primary immunodeficiency diseases: single center experience in Jordan. Pediatr Transplant. 2013;17(4):394–402 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23692601.
Bacalhau S, Freitas C, Valente R, Barata D, Neves C, Schäfer K, et al. Successful handling of disseminated BCG disease in a child with severe combined immunodeficiency. Case Rep Med. 2011;2011:527569 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22110512.
Bernatowska EA, Wolska-Kusnierz B, Pac M, Kurenko-Deptuch M, Zwolska Z, Casanova JL, et al. Disseminated bacillus Calmette-Guérin infection and immunodeficiency [14]. Emerg Infect Dis. Centers for Disease Control and Prevention (CDC). 2007;13:799–801.
Heyderman RS, Morgan G, Levinsky RJ, Strobel S. Successful bone marrow transplantation and treatment of BCG infection in two patients with severe combined immunodeficiency. Eur J Pediatr. 1991;150(7):477–80.
Ikincioğullari A, Doğu F, Ciftci E, Unal E, Ertem M, Reisli I, et al. An intensive approach to the treatment of disseminated BCG infection in a SCID patient. Bone Marrow Transplant. 2002;30(1):45–7 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12105777.
Laberko A, Yukhacheva D, Rodina Y, Abramov D, Konovalov D, Radygina S, et al. BCG-related inflammatory syndromes in severe combined immunodeficiency after TCRαβ+/CD19+ depleted HSCT. J Clin Immunol. 2020;40(4):625–36 [Internet]. [cited 2020 Aug 16]. Available from: https://pubmed.ncbi.nlm.nih.gov/32377975/.
Jaing T-H, Lee W-I, Lin T-Y, Huang J-L, Chen S-H, Chow R. Successful unrelated mismatched cord blood transplantation in an infant with severe combined immunodeficiency and Mycobacterium bovis bacillus Calmette-Guèrin disease. Pediatr Transplant. 2006;10(4):501–4 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16712611.
McKenzie RH, Roux P. Disseminated BCG infection following bone marrow transplantation for X-linked severe combined immunodeficiency. Pediatr Dermatol. 2000;17(3):208–12 [Internet]. [cited 2020 may 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10886754.
Skinner R, Appleton AL, Sprott MS, Barer MR, Magee JG, Darbyshire PJ, et al. Disseminated BCG infection in severe combined immunodeficiency presenting with severe anaemia and associated with gross hypersplenism after bone marrow transplantation. Bone Marrow Transplant. 1996;17(5):877–80 [Internet]. [cited 2020 may 12]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8733714.
Fisch P, Millner M, Müller SM, Wahn U, Friedrich W, Renz H. Expansion of γδt cells in an infant with severe combined immunodeficiency syndrome after disseminated BCG infection and bone marrow transplantation. J Allergy Clin Immunol. 1999;103(6):1218–9.
Lee PPW, Chan KW, Chen TX, Jiang LP, Wang XC, Zeng HS, et al. Molecular diagnosis of severe combined immunodeficiency—identification of IL2RG, JAK3, IL7R, DCLRE1C, RAG1, and RAG2 mutations in a cohort of Chinese and Southeast Asian children. J Clin Immunol. 2011;31(2):281–96 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21184155.
Naselli A, Losurdo G, Giardino S, Morreale G, Savioli C, Castagnola E. Successful management of cutaneous BCG dissemination in a child affected by SCID and receiving allogeneic hematopoietic stem cell transplant. Bone Marrow Transplant. 2015;50(12):1572–3 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26121110.
Wahadneh A, Bin-Dahman H, Habahbeh Z, Abu-Shukear M, Ajarmeh M, Zyood R, Khaled A. Successful second bone marrow transplantation in Omenn's syndrome after bone marrow aplasia: a case report. Pediatr Transplant. 2012;16(2):E43–8. https://doi.org/10.1111/j.1399-3046.2010.01413.x.
Yildiran A, Fişgin T, Güner SN, Kiliç M, Sancak R, Özyürek E, et al. Hemophagocytic bone marrow aplasia with plasma cells in a RAG2-deficient SCID case after a nonconditioned transplantation from a fully matched sibling. J Pediatr Hematol Oncol. 2013;35(8):e335–7 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23389499.
Deffert C, Schäppi MG, Pache JC, Cachat J, Vesin D, Bisig R, Ma Mulone X, Kelkka T, Holmdahl R, Garcia I, Olleros ML, Krause KH. Bacillus calmette-guerin infection in NADPH oxidase deficiency: defective mycobacterial sequestration and granuloma formation. PLoS Pathog. 2014;10(9):e1004325. https://doi.org/10.1371/journal.ppat.1004325.
Lin CJ, Wang SC, Ku CL, Kao JK, Chen M, Liu CS. Successful unrelated cord blood stem cell transplantation in an X-linked chronic granulomatous disease patient with disseminated BCG-induced infection. Pediatr Neonatol. 2015;56(5):346–50.
Moliva JI, Turner J, Torrelles JB. Immune Responses to Bacillus Calmette-Guérin vaccination: why do they fail to protect against Mycobacterium tuberculosis? Front Immunol. 2017;8(APR):407. [Internet]. [cited 2020 APR 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28424703.
Mazzucchelli JTL, Bonfim C, Castro GG, Condino-Neto AA, Costa NMX, Cunha L, et al. Severe combined immunodeficiency in Brazil: management, prognosis, and BCG-associated complications. J Investig Allergol Clin Immunol. 2014;24(3):184–91.
French MA. Immune reconstitution inflammatory syndrome: a reappraisal. Clin Infect Dis. 2009;48(1):101–7 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19025493.
Bozzano F, Marras F, De Maria A. Immunology of tuberculosis [Internet]. Mediterr J Hematol Infect Dis. 2014;6:e2014027 Universita Cattolica del Sacro Cuore; [cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24804000.
Millman AC, Salman M, Dayaram YK, Connell ND, Venketaraman V. Natural killer cells, glutathione, cytokines, and innate immunity against Mycobacterium tuberculosis. J Interf Cytokine Res. 2008 Mar 1;28(3):153–65.
Cotton MF, Rabie H, Nemes E, Mujuru H, Bobat R, Njau B, Violari A, Mave V, Mitchell C, Oleske J, Zimmer B, Varghese G, Pahwa S; P1073 team. A prospective study of the immune reconstitution inflammatory syndrome (IRIS) in HIV-infected children from high prevalence countries. PLoS One. 2019;14(7):e0211155. https://doi.org/10.1371/journal.pone.0211155.
Sun HY, Singh N. Immune reconstitution inflammatory syndrome in non-HIV immunocompromised patients. Curr Opin Infect Dis. 2009;22:394–402.
Scharschmidt TC, Amerson EH, Rosenberg OS, Jacobs RA, McCalmont TH, Shinkai K. Immune reconstitution reactions in human immunodeficiency virus-negative patients: report of a case and review of the literature. JAMA Dermatol. 2013;149(1):74–8.
Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. 2008;122(6):1082–6.
Sun HY, Singh N. Opportunistic infection-associated immune reconstitution syndrome in transplant recipients. Clin Infect Dis. 2011;53(2):168–76 [Internet]. [cited 2020 May 8]. Available from: https://www-ncbi-nlm-nih-gov.clalit-ez.medlcp.tau.ac.il/pubmed/?term=Opportunistic+Infection-Associated+Immune+Reconstitution+Syndrome+in+Transplant+Recipients.
Searle E, Patel H, Vilar FJ, Gharib M, Turner AJ, Batra G, et al. Inflammatory BCG adenitis associated with immune reconstitution following allogeneic haematopoietic stem cell transplant in infancy. Pediatr Blood Cancer. 2010;54(1):166–9 [Internet]. [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19606456.
Yu R, Satoh T, Wakabayashi T, Ueda N, Yokozeki H. Disseminated BCG infection in severe combined immunodeficiency [Internet]. Acta Derm Venereol. 2012;92:158–9 [cited 2020 Apr 16]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21879238.
World Health Organization. BCG vaccine: WHO position paper, February 2018, Recommendations [Internet]. Vaccine. 2018;36:3408–10 Elsevier Ltd; [cited 2020 May 6]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29609965.
Koppel A, Leonardo-Guerrero J, Rives S, Paniagua-Torres N, Sparrow C, Beck-Sagué CM. Immune reconstitution inflammatory syndrome due to Mycobacterium bovis Bacillus Calmette-Guerin in infants receiving highly active antiretroviral therapy: a call for universal perinatal rapid HIV testing prior to administration of BCG immunization of neonates. J Trop Pediatr. 2010;56(4):280–3 [Internet]. [cited 2020 May 14]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19952057.
de Jesus Nunes-Santos C, Rosenzweig SD. Bacille Calmette-Guerin complications in newly described primary immunodeficiency diseases: 2010-2017 [Internet]. Front Immunol. 2018;9:1423 Frontiers Media S.A.[cited 2020 Apr 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29988375.
Bacille Calmette-Guérin (BCG) vaccination and COVID-19 Scientific brief 12 April 2020 Summary. In: clinicaltrials.gov. 2020 [cited 2020; Available from: http://www.clinicaltrials.gov/ct2/show/NCT04327206.
Hamiel U, Kozer E, Youngster I. SARS-CoV-2 Rates in BCG-Vaccinated and Unvaccinated Young Adults. JAMA. 2020;323(22):2340–2341. https://doi.org/10.1001/jama.2020.8189.
Acknowledgments
We thank our patients and their families, and our departmental nursing and administrative staff for their tireless commitment to patient care. Particularly, we thank Mrs. Maram Shweiki for her tireless work which enables optimal patient care. We also thank Professor Zeev Rotstein, Director of The Hadassah Medical Center, for his support of the department and our patients.
Funding
Adeeb NaserEddin received “Award for Excellence 2018” from Allergists For Israel organization. Bella Shadur’s position is supported by the Australian Government Research Training Program Scholarship and by Hadassah Australia. This work was supported by the Deutsche Forschungsgemeinschaft (Discovery and Evaluation of new Combined Immunodeficiency Disease Entities (DECIDE); grant DFG WA 1597/4-2) and the ERA-NetERARE Consortium EURO-CID.
Author information
Authors and Affiliations
Contributions
All authors have read and approved this manuscript. Yael Dinur Schejter, Adeeb NaserEddin, and Bella Shadur gathered the data, treated the patients, and wrote the manuscript. Oded Shamriz, Yuval Tal, Irina Zaidman, Ehud EvenOr, and Diana Averbuch gathered data and treated patients. Klaus Warnatz assisted in writing, assembly, and editing. Avraham Shaag and Orly Elpeleg provide the genetic diagnosis and segregation studies. Polina Stepensky treated the patients, designed, and supervised the study.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
NaserEddin, A., Dinur-Schejter, Y., Shadur, B. et al. Bacillus Calmette–Guerin (BCG) Vaccine-associated Complications in Immunodeficient Patients Following Stem Cell Transplantation. J Clin Immunol 41, 147–162 (2021). https://doi.org/10.1007/s10875-020-00892-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-020-00892-6