
Abstract
Advanced NMR methods combined with biophysical techniques have recently provided unprecedented insight into structure and dynamics of molecular chaperones and their interaction with client proteins. These studies showed that several molecular chaperones are able to dissolve aggregation-prone polypeptides in aqueous solution. Furthermore, chaperone-bound clients often feature fluid-like backbone dynamics and chaperones have a denaturing effect on clients. Interestingly, these effects that chaperones have on client proteins resemble the effects of known chaotropic substances. Following this analogy, chaotropicity could be a fruitful concept to describe, quantify and rationalize molecular chaperone function. In addition, the observations raise the possibility that at least some molecular chaperones might share functional similarities with chaotropes. We discuss these concepts and outline future research in this direction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Atomic resolution studies of chaperone–client systems
Cells in all kingdoms of life possess helper proteins—the chaperones—to perform essential tasks in the genesis of biomacromolecular structure (Ellis 1993; Bukau et al. 2006; Horwich and Fenton 2009; Hartl et al.. 2011; Georgescauld et al. 2014; Schopf et al. 2017). Chaperones can be classified into two groups, based on the broadness of their substrate range, the clientome. “Specialized chaperones” are specific for a single client or a few clients, while “general chaperones” have large clientomes with up to 100s of members (Bose and Chakrabarti 2017). The functionality of general chaperones is typically transitive among homologues, across species and even between different chaperone types, such that their clientomes overlap at least partially. In vitro assays for chaperone activity, such as the prevention of aggregation of model proteins, frequently show that different general chaperones function similarly, including ATP-independent as well as ATP-dependent ones (Stenberg and Fersht 1997; Gray and Fersht 1993; Gray et al. 1993; Entzminger et al. 2012; Huang et al. 2016; Burmann et al. 2020). Overall, these notions suggest that common biophysical principles are shared between general chaperones.
The best characterized general chaperones are arguable the ATP-dependent Hsp90, Hsp60 and Hsp70 from the Heat shock protein (Hsp) family (Schlecht et al. 2011; Barducci and De Los Rios 2015; Sontag et al. 2017; Wruck et al. 2018; Burmann et al. 2020; Sousa et al. 2016; Goloubinoff et al. 2018; Rebeaud et al. 2020; Schopf et al. 2017). These large molecular machines integrate with several cofactors (co-chaperones) towards dedicated functional cycles, often in a modular fashion. Other general chaperones of high biological interest include the ATP-independent prefoldin, trigger factor, SecB, and the bacterial periplasmic chaperones SurA, Spy and Skp. These chaperones feature the “holdase” function—the ability to bind unfolded and partially folded clients for extended time periods.
As for all biomacromolecules, resolving structural and functional features at atomic resolution is key to understanding the biological function. For many of the key chaperones, atomic structures of the client-free forms have long been available (Braig et al. 1994; Xu et al. 2000; Bitto and McKay 2002; Ferbitz et al. 2004; Korndörfer et al. 2004; Walton and Sousa 2004; Webb et al. 2006; Ali et al. 2006; Zhang et al. 2010; Kityk et al. 2012). In contrast, structural descriptions of client-interacting states have long escaped experimental access, arguably so, because chaperone-bound clients are highly dynamic and frequently adopt multi-conformational ensembles, rendering them unsuitable for coherent averaging of diffraction or transmission data (Hiller and Burmann 2018). For example, cryo-electron microscopy could detect client proteins bound to Hsp60 or Hsp90 only at low resolution, preventing determination of atomic coordinates (Clare et al. 2009; Chen et al. 2013; Verba et al. 2016; Cuéllar et al. 2019).
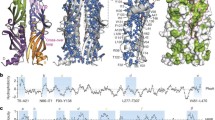
The situation changed substantially in the early 2010s with the success of modern high-resolution techniques of NMR spectroscopy. Advanced isotope labelling combined with spectroscopic techniques including TROSY had raised the size limitations for functional and structural studies of biomolecular systems towards the hundreds of kDa, in particular for systems with a known ground structure (Pervushin et al. 1997; Tugarinov et al. 2003; Sprangers et al. 2007; Mas et al. 2013; Goto et al. 1999). When combined with advanced biochemical preparations, these techniques thus enabled the first complete description of a full-length client in complex with a chaperone, the Omp–Skp system (Burmann et al. 2013) (Fig. 1a). In the following years, client-bound states of multiple chaperones were resolved to atomic level, including trigger factor, SecB, Tim9/10, Spy, Hsp40, Hsp70 and Hsp90 (Saio et al. 2014; Karagöz et al. 2014; Huang et al. 2016; Salmon et al. 2016; He et al. 2016; Rosenzweig et al. 2017; Weinhäupl et al. 2018; Jiang et al. 2019) (Fig. 1b–d). In many of these cases, the client proteins bound to the chaperone adopt dynamic ensemble states. Notably, the dynamic interconversion of conformations while remaining bound to the chaperone is in full agreement with a number of functional studies (Gray and Fersht 1993; Gray et al. 1993; Stenberg and Fersht 1997; Burmann et al. 2013; Thoma et al. 2015; He et al. 2016; Stull et al. 2016; Horowitz et al. 2017; Hiller and Burmann 2018), highlighting client fluidity as a central feature for many chaperones and the need to describe them by ensembles rather than static structures. Additional notable NMR studies yielded valuable insights into further functional aspects of general chaperones, including the Hsp60 system (TRiC/CCT (Joachimiak et al. 2014), GroEL (Libich et al. 2015, 2017) and thermosome (Mas et al. 2018)), Hsp70 (Zhuravleva et al. 2012; Libich et al. 2013; Rosenzweig et al. 2013; Sekhar et al. 2015), and Hsp90 (Park et al. 2011; Oroz et al. 2017). Overall, from these high-resolution studies, common features emerged that connect the functionality of several chaperones. We discuss these two of these features in the following.

Dynamic ensemble models of general chaperones in complex with clients. a Bacterial chaperone Skp (yellow) with the bound client protein OmpX (purple) (Burmann et al. 2013). b Conformational ensemble of the intrinsically disordered protein Tau (yellow) bound to chaperone Hsp90 (cyan/green) (Karagöz et al. 2014). c, d Chaperone Spy (grey) with bound client Im7 (c) or SH3 (d) (He et al. 2016; He and Hiller 2018, 2019). Different conformations of the bound client are labeled by different colors. See original publications for all details. Reproduced with permissions
Solubilization of aggregation-prone polypeptides
The first common feature is the ability to solubilize aggregation-prone polypetides that would otherwise readily precipitate in aqueous solution. Among other, this function is performed by the periplasmic chaperone Skp, that solubilizes unfolded outer membrane proteins (Omps) in chemical equilibrium. In the absence of the chaperone, the Omps immediately precipitate in the same aqueous buffer (Burmann et al. 2013; Schiffrin et al. 2016). Thereby, the NMR spectra of the Omp clients bound to the chaperone feature a narrow chemical shift dispersion for all resonances, both in the backbone and the side chains (Fig. 2b). This indicates random-like sampling of backbone dihedral angles in the Ramachandran space with fast kinetics (Burmann et al. 2013; Callon et al. 2014). The presence of fast interconversion dynamics in the clients was further quantified by NMR measurements (Burmann et al. 2013). The backbone dynamics of the Skp-bound clients OmpX and tOmpA on the ps–ns timescale, as described by the spectral density function J(ωN) and J(0.87ωH), resembles in magnitude the values that the same polypeptide has in 8 M urea solution, indicating high local flexibility (Fig. 2a). In contrast, the dynamics J(0), which correspond to the effective overall molecular tumbling, are strongly elevated compared to the urea-denatured form, reflecting the coupling of the Omp client to the large molecular weight of the chaperone. Notably, despite the fast local dynamics, the chaperone-bound state is spatially compacted relative to the random coil ensemble in the bulk chaotrope urea (Fig. 2c).

Similarities of chaperone- and urea-solubilized client states. a Residue-specific backbone dynamics of tOmpA bound to chaperone Skp. The spectral density function J(ω) is evaluated at three timescales. The average values bound to Skp is indicated by a black dashed line and in 8 M urea by an orange dashed line. b Similarity of 2D [15N,1H]-TROSY spectra of OmpX in chaperone and in urea. c Compactness of the OmpX polypeptide in 8 M urea solution (left) and bound to chaperone Skp (right) as determined by intramolecular PRE. Low Vox/Vred values indicate special proximity of the residue to the paramagnetic probe at residue 140. A single model conformation of the polypeptide is shown below for each case and the corresponding radius of gyration r is indicated. See original publications for all details.
Compaction of the client and fast local polypeptide dynamics of a chaperone-bound client was also observed for the ATP-independent chaperone Spy interacting with the soluble client protein Im7M. Spy compacts the disordered client, but keeps it in an overall dynamic state that reorients on the chaperone surface (He et al. 2016; Stull et al. 2016). For three other molecular chaperones, trigger factor, SurA, and SecB, spectra of the bound outer membrane protein client showed similarly narrow chemical shift dispersion. Just as for the chaperone Skp, these spectra indicate indicating fast averaging of backbone dihedral angles and demonstrate that these chaperones also solubilize the Omp clients for extended time periods in fluidic states (Fig. 3). Taken together, the five chaperones trigger factor, Skp, SurA, SecB and Spy are able to dissolve aggregation-prone polypeptides in aqueous solution in thermal equilibrium.

Reproduced from (Burmann et al. 2013) with permission
Different chaperones solubilize the membrane protein tOmpA in an unfolded state. 2D [15N,1H]-TROSY NMR fingerprint spectra of [U-2H,15N]-tOmpA bound to each of the unlabeled chaperones SecB (green), trigger factor (TF, purple), SurA (orange) and Skp (blue). All spectra were recorded at 37 °C in aqueous buffer. The PDB structure of the respective chaperone is shown above each spectrum, indicating their structural diversity. Note that in the absence of chaperone, tOmpA is not soluble in the same buffer.
Denaturation of folded and partially folded clients
A second common functional theme of chaperones that was observed in multiple studies is a denaturing effect on client proteins. In one exemplary work, the effect of the ATP-independent periplasmic chaperone Spy on the partially folded client protein Im7 was characterized by high-resolution NMR spectroscopy (He et al. 2016). Spy was found to interact with Im7 via a specific local region of the client protein and this interaction was shown to induce unfolding of the entire protein. Several spectroscopic parameters evidence this denaturation effect: The local dynamics of the polypeptide on the ps–ns timescale was globally increased, as measured by the heteronuclear NOE (Fig. 4a). Secondary chemical shifts, which are a direct measure for helical secondary structure propensity, were also reduced in the entire protein (Fig. 4b). And thirdly, the kinetic exchange rates of amide protons with water were significantly increased in the entire domain upon interaction with the chaperone (Fig. 4c, d). These data thus indicate that the conformational equilibrium between folded and unfolded Im7 is shifted towards the unfolded conformation by the interaction with the chaperone Spy, corresponding to a denaturation.

Denaturation of proteins by molecular chaperones. a–d Effect of the chaperone Spy on the client protein Im7 (He et al. 2016). a Difference in heteronuclear NOE of the client upon chaperone binding. A positive value means an increase in local flexibility. b Difference in secondary chemical shifts. Positive values indicate a decrease in helical propensity. c, d Backbone amide proton exchange in absence (c, grey) and presence (d, orange) of Spy. e Effect of the chaperone hsp60 on the melting temperature of the client protein HEWL (Mas et al. 2018). The population of folded and unfolded protein has been quantified as a function of temperature using NMR spectroscopy. See original works for all details.
The equivalent effect was observed in several independent studies also for very different molecular chaperones, the ATP-dependent Hsp60 family. Early studies using H/D exchange evidenced an unfolding activity of the bacterial Hsp60 variant GroEL on the substrate Rubisco, notably in the absence of ATP or any other source of Gibbs free energy (Shtilerman et al. 1999). The same observation was subsequently also made for other clients, for both GroEL and the Hsp60 form CCT (Priya et al. 2013a, b). More recently, solution NMR spectroscopic observations of the model client protein HEWL (hen egg white lysozyme), showed that archaeal Hsp60 decreased its melting temperature by about five degrees, effectively denaturing the client protein (Mas et al. 2018) (Fig. 3e).
Also for the large chaperone Hsp70, the specific activity to unfold client proteins has been repeatedly reported. When embedded into functional cycles, such unfoldase function of Hsp70 eventually leads to an effective folding reaction for clients, but the elementary underlying function of Hsp70 was shown to unfold rather than to fold the clients (Ben-Zvi et al. 2004; Morán Luengo et al. 2018, 2019). Additional observations of client unfolding activity have also been reported also for Hsp90 and for the small heat-shock proteins (sHsps) (Finka et al. 2016). Overall, these multiple observations evidence a denaturation effect for a majority of the general chaperones, including the most prominent members Hsp60, Hsp70 and Hsp90, as well as multiple classical holdases.
Chaotropic compounds and their effect on proteins
The terminology of “chaotropicity” was introduced in 1962 to classify compounds that destabilize biomolecular structures in aqueous solution, i.e. decrease their melting temperature (Hamaguchi and Geiduschek 1962). Contemporary lists of chaotropes contain over 60 small molecule substances, including also the Hofmeister series, one of the earliest collections of compounds that modulate the solubility of proteins (Hofmeister 1888; Baldwin 1996; Zhang and Cremer 2009; Cray et al. 2013). In aqueous solutions of chaotropes, the unfolded–folded equilibrium is shifted towards the unfolded state, leading to an effective destabilization of biomacromolecular structure. Notably, for a given chaotrope this effect is not specific to certain targets, but acts in a non-specific fashion on a wide range of biomacromolecules. At the same time, the effect of a chaotrope on a given target protein is non-uniform, as it depends on the presence and the features of local structure. Structured regions are being unfolded, while unstructured regions essentially remain as before. The mechanism of chaotrope action remains puzzling despite intensively being studied (Ball and Hallsworth 2015). Chaotropes have been proposed as perturbers of water structure and thus de-stabilizers of protein structure (Ball and Hallsworth 2015). Thereby, it is not yet clear whether the effects chaotropes have on water are an unavoidable consequence of the effects chaotropes have on biomolecules, or whether the water effects are mechanistically required to then lead to the destabilization of the biomacromolecules and whether all chaotropes necessarily act via the same or similar mechanisms (Ball and Hallsworth 2015). Alternative mechanistic suggestions rely on hydrophobic cavity effects (Breslow and Guo 1990; Baldwin 1996; Graziano 2011), or direct interaction with the polypeptide backbone (Möglich et al. 2005). Molecular dynamics simulations reported that the chaotrope urea destabilizes proteins by both indirect and direct mechanisms, i.e. by perturbation of the water structure and interference with the hydrophobic core of the protein, but also by interactions with the polypeptide backbone (Bennion and Daggett 2003). Irrespective of the underlying mechanism, as a net effect, chaotropes dissolve aggregation-prone segments of polypeptides better than aqueous solution alone, notably without necessarily being hydrophobic substances themselves. The dissolved polypeptides adopt random-coil conformations, emerging from independent local sampling of the Ramachandran space at each polypeptide bond (Shortle 1996; Smith et al. 1996). Furthermore, among the large variety of chaotropes, only a limited subset are able to dissolve aggregation-prone proteins such as membrane proteins in aqueous solution. This subset comprises urea, guanidinium, thioisocyanate and perchlorate. While bulk organic solvents can also dissolve polypeptides in unfolded forms, only the above-mentioned set of chaotropes can do this task in aqueous solution. In other words, the ability to dissolve aggregation-prone peptides constitutes a strong identification criterion for chaotropicity.
Comparing molecular chaperones and chaotropes
When summarizing the above-mentioned functional properties of chaperones and chaotropes, certain similarities become apparent. Chaotropes and chaperones are the only known molecules that can solubilize aggregation-prone peptides in aqueous solution, and both types of molecules have a denaturing effect on client proteins. These observations thus suggest that chaotropicity might be a fruitful theoretical concept to describe the function of at least some chaperones.
In addition, the similarities between molecular chaperones and classical chaotropes bear the interesting possibility that at least in some cases, certain underlying mechanistic principles might be shared. For ATP-independent chaperones, chaotropicity might well be correlated with the holdase function, in the sense that free energy gained from the chaotrope effect at least partially contributes to the binding affinity between chaperone and client. For more complex chaperones, chaotropicity might be the underlying basic function that is then embedded in regulatory mechanisms to result in complicated functional cycles. Uncontrolled chaotropicity is generally detrimental to cells and the embedding and protection into functional cycles may in many cases serve the purpose to prevent unwanted damaging effects towards off-targets by tight control. Of further note, the concept of chaotropicity as the molecular basis for chaperone function is in full agreement with findings that both hydrophobic and non-hydrophobic interactions are involved in chaperone function (Qu et al. 2007; He et al. 2016; Koldewey et al. 2016).
The concept to consider chaperones chaotropes may bring interesting perspectives for both chaperone and chaotrope research, in particular, since the individual mechanisms underlying the classical chaotropes are also still not fully understood (Ball and Hallsworth 2015). Atomic resolution studies of chaperones and additional biophysical measurements may thus also contribute to better understand the mechanisms underlying classical chaotropes. Because none of the 20 proteinogenic amino acids is itself a chaotrope, evolution appears to have created chaotropicity by suitable spatial arrangement of non-chaotropic elements. We anticipate that combinations of experiments with bioinformatic analyses will eventually reveal the molecular principles for the creation of chaotropic surfaces in chaperones. Along these lines, it should become possible to define a quantitative measure for chaotropicity of chaperones and connect this to classical chaotropes.
References
Ali MM et al (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440:1013–1017
Baldwin RL (1996) How Hofmeister ion interactions affect protein stability. Biophys J 71:2056–2063
Ball P, Hallsworth JE (2015) Water structure and chaotropicity: their uses, abuses and biological implications. Phys Chem Chem Phys 17:8297–8305
Barducci A, De Los RP (2015) Non-equilibrium conformational dynamics in the function of molecular chaperones. Curr Opin Struct Biol 30:161–169
Bennion BJ, Daggett V (2003) The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci USA 100:5142–5147
Ben-Zvi A, De Los RP, Dietler G, Goloubinoff P (2004) Active solubilization and refolding of stable protein aggregates by cooperative unfolding action of individual hsp70 chaperones. J Biol Chem 279:37298–37303
Bitto E, McKay DB (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10:1489–1498
Bose D, Chakrabarti A (2017) Substrate specificity in the context of molecular chaperones. IUBMB Life 69:647–659
Braig K et al (1994) The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature 371:578–586
Breslow R, Guo T (1990) Surface tension measurements show that chaotropic salting-in denaturants are not just water-structure breakers. Proc Natl Acad Sci USA 87:167–169
Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125:443–451
Burmann BM, Wang C, Hiller S (2013) Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat Struct Mol Biol 20:1265–1272
Burmann BM et al (2020) Regulation of α-synuclein by chaperones in mammalian cells. Nature 577:127–132
Callon M, Burmann BM, Hiller S (2014) Structural mapping of a chaperone-substrate interaction surface. Angew Chem Int Ed Engl 53:5069–5072
Chen DH et al (2013) Visualizing GroEL/ES in the act of encapsulating a folding protein. Cell 153:1354–1365
Clare DK et al (2009) Chaperonin complex with a newly folded protein encapsulated in the folding chamber. Nature 457:107–110
Cray JA et al (2013) A universal measure of chaotropicity and kosmotropicity. Environ Microbiol 15:287–296
Cuéllar J et al (2019) Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly. Nat Commun 10:1–14
Ellis RJ (1993) The general concept of molecular chaperones. Phil Trans Royal Soc London B 339:257–261
Entzminger KC et al (2012) The Skp chaperone helps fold soluble proteins in vitro by inhibiting aggregation. Biochemistry 51:4822–4834
Ferbitz L et al (2004) Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 431:590–596
Finka A, Mattoo RU, Goloubinoff P (2016) Experimental milestones in the discovery of molecular chaperones as polypeptide unfolding enzymes. Annu Rev Biochem 85:715–742
Georgescauld F et al (2014) GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM-barrel domain folding. Cell 157:922–934
Goloubinoff P et al (2018) Chaperones convert the energy from ATP into the nonequilibrium stabilization of native proteins. Nat Chem Biol 14:388–395
Goto NK et al (1999) A robust and cost-effective method for the production of Val, Leu, Ile (d1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR 13:369–374
Gray TE, Fersht AR (1993) Refolding of barnase in the presence of GroE. J Mol Biol 232:1197–1207
Gray TE et al (1993) Refolding of barnase mutants and pro-barnase in the presence and absence of GroEL. EMBO J 12:4145–4150
Graziano G (2011) Contrasting the denaturing effect of guanidinium chloride with the stabilizing effect of guanidinium sulfate. Phys Chem Chem Phys 13:12008–12014
Hamaguchi K, Geiduschek EP (1962) The effect of electrolytes on the stability of the deoxyribonucleate Helix. J Am Chem Soc 84:1329–1338
Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475:324–332
He L, Hiller S (2018) Common patterns in chaperone interactions with a native client protein. Angew Chem Int Ed Engl 57:5921–5924
He L, Hiller S (2019) Frustrated interfaces facilitate dynamic interactions between native client proteins and holdase chaperones. ChemBioChem 20:2803–2806
He L, Sharpe T, Mazur A, Hiller S (2016) A molecular mechanism of chaperone-client recognition. Sci Adv 2:e1601625
Hiller S, Burmann BM (2018) Chaperone-client complexes: a dynamic liaison. J Magn Reson 289:142–155
Hofmeister F (1888) Zur Lehre von der Wirkung der Salze. II. Arch Exp Pathol Pharmakol 24:247–260
Horowitz S, Koldewey P, Stull F, Bardwell JC (2017) Folding while bound to chaperones. Curr Opin Struct Biol 48:1–5
Horwich AL, Fenton WA (2009) Chaperonin-mediated protein folding: using a central cavity to kinetically assist polypeptide chain folding. Q Rev Biophys 42:83–116
Huang C, Rossi P, Saio T, Kalodimos CG (2016) Structural basis for the antifolding activity of a molecular chaperone. Nature 537:202–206
Jiang Y, Rossi P, Kalodimos CG (2019) Structural basis for client recognition and activity of Hsp40 chaperones. Science 365:1313–1319
Joachimiak LA et al (2014) The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159:1042–1055
Karagöz GE et al (2014) Hsp90-tau complex reveals molecular basis for specificity in chaperone action. Cell 156:963–974
Kityk R, Kopp J, Sinning I, Mayer MP (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell 48:863–874
Koldewey P et al (2016) Forces driving chaperone action. Cell 166:369–379
Korndörfer IP, Dommel MK, Skerra A (2004) Structure of the periplasmic chaperone Skp suggests functional similarity with cytosolic chaperones despite differing architecture. Nat Struct Mol Biol 11:1015–1020
Libich DS, Fawzi NL, Ying J, Clore GM (2013) Probing the transient dark state of substrate binding to GroEL by relaxation-based solution NMR. Proc Natl Acad Sci USA 110:11361–11366
Libich DS, Tugarinov V, Clore GM (2015) Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc Natl Acad Sci USA 112:8817–8823
Libich DS, Tugarinov V, Ghirlando R, Clore GM (2017) Confinement and stabilization of Fyn SH3 folding intermediate mimetics within the cavity of the chaperonin GroEL demonstrated by relaxation-based NMR. Biochemistry 56:903–906
Mas G et al (2013) Specific labeling and assignment strategies of valine methyl groups for NMR studies of high molecular weight proteins. J Biomol NMR 57:251–262
Mas G et al (2018) Structural investigation of a chaperonin in action reveals how nucleotide binding regulates the functional cycle. Sci Adv 4:eaau4196
Möglich A, Krieger F, Kiefhaber T (2005) Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J Mol Biol 345:153–162
Morán Luengo ML, Kityk R, Mayer MP, Rüdiger R (2018) Hsp90 breaks the deadlock of the Hsp70 chaperone system. Mol Cell 70:545–552
Morán Luengo ML, Mayer MP, Rüdiger R (2019) The Hsp70-Hsp90 chaperone cascade in protein folding. Trends Cell Biol 29:164–177
Oroz J, Kim JH, Chang BJ, Zweckstetter M (2017) Mechanistic basis for the recognition of a misfolded protein by the molecular chaperone Hsp90. Nat Struct Mol Biol 24:407–413
Park SJ, Borin BN, Martinez-Yamout MA, Dyson HJ (2011) The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat Struct Mol Biol 18:537–541
Pervushin K, Riek R, Wider G, Wüthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole—dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371
Priya S, Sharma SK, Goloubinoff P (2013) Molecular chaperones as enzymes that catalytically unfold misfolded polypeptides. FEBS Lett 587:1981–1987
Priya S et al (2013) GroEL and CCT are catalytic unfoldases mediating out-of-cage polypeptide refolding without ATP. Proc Natl Acad Sci USA 110:7199–7204
Qu J et al (2007) The trimeric periplasmic chaperone Skp of Escherichia coli forms 1:1 complexes with outer membrane proteins via hydrophobic and electrostatic interactions. J Mol Biol 374:91–105
Rebeaud ME, Mallik S, Goloubinoff P, Tawfik DS (2020) On the evolution of chaperones and co-chaperones and the exponential expansion of proteome complexity. bioRxiv. 2020.06.08.140319
Rosenzweig R et al (2013) Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science 339:1080–1083
Rosenzweig R, Sekhar A, Nagesh J, Kay LE (2017) Promiscuous binding by Hsp70 results in conformational heterogeneity and fuzzy chaperone-substrate ensembles. Elife 6:1–22
Saio T et al (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344:1250494
Salmon L et al (2016) Capturing a dynamic chaperone-substrate interaction using NMR-informed molecular modeling. J Am Chem Soc 138:9826–9839
Schiffrin B et al (2016) Skp is a multivalent chaperone of outer-membrane proteins. Nat Struct Mol Biol 23:786–793
Schlecht R, Erbse AH, Bukau B, Mayer MP (2011) Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol 18:345–351
Schopf FH, Biebl MM, Buchner J (2017) The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18:345–360
Sekhar A, Rosenzweig R, Bouvignies G, Kay LE (2015) Mapping the conformation of a client protein through the Hsp70 functional cycle. Proc Natl Acad Sci USA 112:10395–10400
Shortle D (1996) The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J 10:27–34
Shtilerman M, Lorimer GH, Englander SW (1999) Chaperonin function: folding by forced unfolding. Science 284:822–825
Smith LJ, Fiebig KM, Schwalbe H, Dobson CM (1996) The concept of a random coil. Residual structure in peptides and denatured proteins. Fold Des 1:R95-106
Sontag EM, Samant RS, Frydman J (2017) Mechanisms and functions of spatial protein quality control. Annu Rev Biochem 86:97–122
Sousa R et al (2016) Clathrin-coat disassembly illuminates the mechanisms of Hsp70 force generation. Nat Struct Mol Biol 23:821–829
Sprangers R, Velyvis A, Kay LE (2007) Solution NMR of supramolecular complexes: providing new insights into function. Nat Methods 4:697–703
Stenberg G, Fersht AR (1997) Folding of barnase in the presence of the molecular chaperone SecB. J Mol Biol 274:268–275
Stull F et al (2016) Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat Struct Mol Biol 23:53–58
Tafer H et al (2004) Nonrandom structure in the urea-unfolded Escherichia coli outer membrane protein X (OmpX). Biochemistry 43:860–869
Thoma J, Burmann BM, Hiller S, Müller DJ (2015) Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat Struct Mol Biol 22:795–802
Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE (2003) Cross-correlated relaxation enhanced 1H[-]13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc 125:10420–10428
Verba KA et al (2016) Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352:1542–1547
Walton TA, Sousa MC (2004) Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 15:367–374
Webb CT et al (2006) Crystal structure of the mitochondrial chaperone TIM9.10 reveals a six-bladed alpha-propeller. Mol Cell 21:123–133
Weinhäupl K et al (2018) Structural basis of membrane protein chaperoning through the mitochondrial intermembrane space. Cell 175:1365–1379
Wruck F et al (2018) Protein folding mediated by Trigger Factor and Hsp70: new insights from single-molecule approaches. J Mol Biol 430:438–449
Xu Z, Knafels JD, Yoshino K (2000) Crystal structure of the bacterial protein export chaperone SecB. Nat Struct Biol 7:1172–1177
Zhang Y, Cremer PS (2009) The inverse and direct Hofmeister series for lysozyme. Proc Natl Acad Sci USA 106:15249–15253
Zhang J et al (2010) Mechanism of folding chamber closure in a group II chaperonin. Nature 463:379–384
Zhuravleva A, Clerico EM, Gierasch LM (2012) An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151:1296–1307
Acknowledgements
Anne Spang, Guillaume Mas, Jakub Macošek, Johanna Ude, Stefan Rüdiger and Thomas Müntener are kindly acknowledged for valuable comments and discussion. I would also like to thank all present and past members of my group that have contributed with their work to our understanding of chaperones.
Funding
Open access funding provided by University of Basel. This work was funded by the Swiss National Science Foundation (310030B_185388), which is particularly endorsed for their continuous support of our research in the past 10 years.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hiller, S. Molecular chaperones and their denaturing effect on client proteins. J Biomol NMR 75, 1–8 (2021). https://doi.org/10.1007/s10858-020-00353-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-020-00353-7