
Abstract
We present here a set of 13C-direct detected NMR experiments to facilitate the resonance assignment of RNA oligonucleotides. Three experiments have been developed: (1) the (H)CC-TOCSY-experiment utilizing a virtual decoupling scheme to assign the intraresidual ribose 13C-spins, (2) the (H)CPC-experiment that correlates each phosphorus with the C4′ nuclei of adjacent nucleotides via J(C,P) couplings and (3) the (H)CPC-CCH-TOCSY-experiment that correlates the phosphorus nuclei with the respective C1′,H1′ ribose signals. The experiments were applied to two RNA hairpin structures. The current set of 13C-direct detected experiments allows direct and unambiguous assignment of the majority of the hetero nuclei and the identification of the individual ribose moieties following their sequential assignment. Thus, 13C-direct detected NMR methods constitute useful complements to the conventional 1H-detected approach for the resonance assignment of oligonucleotides that is often hindered by the limited chemical shift dispersion. The developed methods can also be applied to large deuterated RNAs.
Similar content being viewed by others
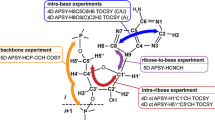

Introduction
The potential of 13C-direct detection in biomolecular NMR has been amply demonstrated (see (Bermel et al. 2006) for review). Initially 13C-direct detection methods were particularly inspired by applications to folded, paramagnetic and deuterated proteins (Serber et al. 2000, 2001; Bermel et al. 2003; Eletsky et al. 2003; Pervushin and Eletsky 2003; Bertini et al. 2004a, b). Concerning applications to RNA oligonucleotides, the groups of Sklenar and Carlomagno have previously introduced pulse sequences based on 13C-direct detection for the resonance assignment of the nucleobase moieties (Fares et al. 2007; Fiala and Sklenar 2007). 13C-direct detected NMR experiments have benefited from the developments in cryogenic probe technology. In particular, the availability of probes optimized for direct low-γ-nuclei detection (Kovacs et al. 2005) has overcome previous sensitivity drawbacks encountered when using proton detection optimized cryoprobes. Although 13C-detection suffers from lower sensitivity compared to 1H-detection, the development of schemes that utilize carbon direct detection offers several advantages. For NMR experiments applied to larger molecules, 13C-direct detection schemes can, for example, reduce the number of coherence transfer modules leading to shorter pulse sequences. Detection of non-protonated carbons including quaternary as well as deuterated carbons becomes feasible, and evolution of 13C chemical shifts in the direct time domain exploits the favourable large 13C chemical shift dispersion without the need to increase sampling in the indirect dimension. Low-γ-nuclei detection also leads to higher salt tolerance and, on a more pragmatic side, the absence of dominant solvent or buffer signals can improve baseline quality of the spectra and reduce t1-noise (Shimba et al. 2004). These advantages make 13C-direct detected experiments a potentially useful complement to the conventional proton detection. By contrast, a potential drawback of carbon direct detection is the wide range of 13C T1-times. Thus, optimized excitation schemes including low flip angle excitation together with band selective excitation become interesting optimization parameters (Felli et al. 2009) in 13C-direct detected experiments. In addition, pulse sequences have to be optimized in order to remove 1J(C,C) couplings in indirect and direct time dimensions.
Here, we report the development of three 13C-direct detected NMR-experiments that correlate 13C- and/or 31P-nuclei to obtain the heteronuclear resonance assignment of the intraresidual ribose carbon nuclei and the phosphorus nuclei along the phosphodiester backbone in 13C-labelled RNAs. These experiments can also directly be applied to 13C-labelled DNAs. We have developed the pulse sequences using two different RNAs spanning a factor of two in molecular weight (14mer and 27mer RNA hairpin structures). We further show the general applicability to larger RNAs by using a selectively 13C-labelled 70mer riboswitch RNA. For such larger RNAs, the introduction of deuterium labelling might generally be beneficial; the implementation of deuterium decoupling into the pulse sequences, introduced here, is straight forward and will allow NMR resonance assignment experiments to be applied for completely deuterated RNAs or for RNAs with specific 1H/2H-labelling schemes in the ribosyl moiety.
In RNA, the intrinsic low chemical shift dispersion of the ribose protons turns their NMR assignment into a considerable challenge. In particular, the sequential assignment might be hindered by the fact that robust NOE-based assignment strategies can only reliably be applied in canonical RNA structural motifs. In these helical elements, the sequential assignment is mainly based on the observation of imino–imino NOE connectivities between successive base pairs which benefit from advantageous spectral resolution (Fürtig et al. 2003). In the cases of unusual secondary and tertiary structure such approaches often fail. The NOE-based assignment of RNAs is further complicated since many labile, nitrogen-bound protons in the nucleobases undergo fast chemical exchange with the solvent and thus cannot be detected. When applying the NOE-based assignment approach, the correct fold, however, cannot be determined when a large number of sequential NOE connectivities are missing due to these fast-exchanging amino or imino protons.
These observations previously led to the development of the HCP and HCP-CCH-TOCSY-experiments (Marino et al. 1994, 1995). Still, restricted chemical shift dispersion of the phosphorus nuclei and strong line broadening due to the large CSA of 31P (Rinnenthal et al. 2009) complicate the analysis of these spectra. In addition, chemical exchange effects often mediated by specific or non-specific binding of mono and divalent cations might be observed on all NMR-active nuclei. The use of 13C-direct detected 13C/13C- and 13C/31P-correlated experiments can facilitate the sequential assignment for the following reasons: (1) the extension of the described pulse sequences to deuterated RNA samples is straight forward, for which the carbon T2-times are considerable longer (Hennig et al. 1997); (2) line broadening of carbon nuclei, especially those induced by divalent ions, can be significantly smaller than of proton nuclei.
In the current study, all carbon nuclei of the ribose moieties (C1′–C5′) and the phosphorus signals of a 13C,15N-labelled 14mer RNA hairpin were assigned using solely the multi-dimensional 13C-direct detected correlation experiments confirming the original resonance assignment (Fürtig et al. 2004). The same set of experiments was applied to a 27mer RNA with selectively 13C-labelled nucleotides (see Fig. 4) (Fürtig et al. 2007) to illustrate their applicability for larger RNAs. In order to eliminate homonuclear 13C-13C couplings in the ribose moieties, the original 13C/13C-TOCSY experiment (Eletsky et al. 2003) was modified by introduction of virtual decoupling schemes (Bermel et al. 2003; Duma et al. 2003). Sequential assignment was obtained for both RNA hairpin structures using the newly developed 2D 13C-direct detected 13C/31P-correlated NMR experiments. These were based on direct correlation of the phosphorus chemical shifts with either C1′, C4′ or C5′ nuclei of the ribose moieties.
Materials and methods
The 13C,15N-labelled 14mer cUUCGg-tetraloop RNA (5′-pppGGCACUUCGGUGCC-3′) was purchased from Silantes GmbH (Munich, Germany). The NMR sample (0.7 mM) was prepared in 20 mM KHPO4 and 0.4 mM EDTA (pH 6.4 in 90% H2O and 10% D2O). 1H chemical shifts were referenced to TSP as an external reference. The complete NMR resonance assignment for 1H, 13C, 15N, and 31P resonances has been published previously (Fürtig et al. 2004). Since the RNA structure and the individual coupling constants have been reported (Nozinovic et al. 2010a, b), coherence transfers could be analyzed efficiently. NMR experiments were carried out on a 600 MHz Bruker NMR spectrometer equipped with a 5 mm z-axis gradient 1H [13C, 31P]-TCI cryogenic probe or a 5 mm z-axis gradient 1H [13C, 15N]-TCI cryogenic probe at 298 K. In addition, experiments were performed on a 950 MHz Bruker NMR spectrometer equipped with a 5 mm z-axis gradient 1H [13C, 15N]-TCI cryogenic probe at 298 or 278 K. The data were processed and analyzed using the program TOPSPIN 2.1 (Bruker BioSpin, Germany).
The synthesis of the selectively 13C-labelled 27mer RNA (5′-ACAGGUUCGCCUGUGUUGCGAACCUGC-3′; bold nucleotides represent residues with 13C-labelled ribose moieties) was carried out by P. Wenter and S. Pitsch (EFP Lausanne, Switzerland) as described previously (Quant et al. 1994; Wenter et al. 2006) (sample concentration 1.2 mM).
13C,15N-labelled UTP was purchased from Silantes GmbH (Munich, Germany). The NMR sample (1 mM) was prepared in 20 mM KHPO4 (pH 6.5 in 100% D2O).
The selectively 13C,15N-cytidine-labelled 70mer RNA corresponding to the aptamer domain of a 2′-deoxyguanosine-dependent riboswitch encoded in Mesoplasma florum (Kim et al. 2007) (5′-pppGGGACUUAUACAGGGUAGCAUAAUGGGCUACUGACCCCGCCUUCAAACCUAUUUGGAGACUAUAAGUCCC-3′, bold nucleotides represent the 13C,15N-labelled cytidine residues) was prepared by in vitro transcription from a linearized DNA template and purified as described previously (Stoldt et al. 1998). The stable RNA-ligand complex is formed upon addition of a threefold molar excess of 2′-deoxyguanosine to the RNA. The final sample concentration was 0.5 mM RNA-ligand complex in 25 mM potassium phosphate, 50 mM KCl, 4 mM MgCl2 at pH 6.2 in 99.98 % D2O. 13C, 15N-labelled CTP was purchased from Silantes GmbH (Munich, Germany), unlabelled rNTPs were purchased from Sigma (Munich, Germany).
The experiments were performed using Bruker NMR spectrometers at field strengths of 600 and 950 MHz. All NMR spectrometers were equipped with 5 mm triple resonance z-axis cryogenic probes. Those cryogenic probes are inverse probes with a cold inner 1H coil and a cold outer coil tuned either for 13C/15N/2H or 13C/31P/2H. In addition, the probes are equipped with cold preamplifiers for the 13C-detection on the outer coil.
Results and discussion
For RNA, the chemical shift assignment does not only constitute the basis for further NMR studies of structure and dynamics, but chemical shifts by themselves carry precious conformational information (Wijmenga and van Buuren 1998; Ebrahimi et al. 2001; Fürtig et al. 2003; Ohlenschläger et al. 2008; Cherepanov et al. 2010). Using canonical coordinates, the sugar pucker as well as the exocyclic torsion around the backbone angle γ can be determined. Phosphorus chemical shifts yield qualitative information about the backbone conformation that is difficult, yet not impossible to obtain otherwise (Richter et al. 2000; Nozinovic et al. 2010a, b). Therefore, obtaining close to complete resonance assignment is important in order to derive high resolution structures of RNAs. For larger RNAs, deuterium labelling can greatly improve the spectral quality (Batey et al. 1996; Vallurupalli et al. 2006; Lu et al. 2010) and information about 13C and 31P chemical shifts may represent the only handle to derive information about local conformations. In the following, we discuss the pulse sequences of the three newly developed experiments.
The 3D-(H)CC-TOCSY-H1′C1′ experiment for the intraresidual assignment of the ribose carbon resonances
The carbon-detected version of the HCC-TOCSY-experiment (Serber et al. 2001) developed for the assignment of protein side chains was one of the first applications of the new generation of 13C-direct detected NMR experiments. Variants of this method were applied to large deuterated proteins (Eletsky et al. 2003) and to selectively protonated proteins for the assignment of the methyl groups (Hu et al. 2006; Jordan et al. 2006). In carbon direct detected experiments, the signal intensity is reduced due to the evolution of the 13C–13C scalar couplings during the acquisition time. Several methods have been proposed to alleviate this problem. Instead of using Fourier transformation in the direct dimension, for example, a maximum entropy reconstruction-deconvolution method (Shimba et al. 2003) has been proposed. In addition, multiple-band-selective 13C-decoupling during acquisition (Bermel et al. 2003; Vögeli et al. 2005) and spin-state selection through IPAP-type excitation (Andersson et al. 1998; Ottiger et al. 1998; Duma et al. 2003; Bertini et al. 2004a, b) or S3E-type excitation (Meissner et al. 1997; Bermel et al. 2005) was applied. Thus far, carbon detected HCC-TOCSY experiments removed the 13C–13C scalar couplings from the spectra by maximum entropy reconstruction-deconvolution. This method, however, can only be applied if the respective coupling constants are similar for all residues, as for example 1J(C′,Cα) ≅ 55 Hz in proteins. In the case of the directly connected carbon nuclei in a RNA ribose moiety, the coupling topology is more complex: the carbon atoms C2′, C3′ and C4′ have two coupling partners each, and the 1J(C,C) coupling constants are not uniform but depend on the ribose conformation as shown in Fig. 1. For the multiple-band-selective 13C-decoupling method (Vögeli et al. 2005), the theoretical increase of 2 in the signal-to-noise ratio cannot be obtained since the dwell time is shared between decoupling and digitization time as previously described in detail (Bermel et al. 2003).

Left Secondary structure of the 14mer cUUCGg-tetraloop RNA. Right1J(C1′C2′) coupling constants [Hz] obtained from a 1H,13C-HSQC with a real time evolution period in the indirect dimension measured on the 14mer RNA. Dashed line indicates the mean value obtained for stem residues; black lines represent the corresponding root mean square deviation. U7 and C8 adopt C2′-endo conformation (Nozinovic et al. 2010a, b) and show considerable differences in 1J(C1′,C2′) coupling constants
Therefore, we followed the principle of spin-state selection utilizing IPAP-type schemes in our RNA experiments (Figs. 2 and 5) comparable to methods applied to carbon direct detected experiments for proteins (Duma et al. 2003; Bertini et al. 2004a, b).

Pulse sequence of the carbon direct detected 3D-(H)CC-TOCSY-H1′C1′ experiment with a virtual decoupling scheme in t3. Narrow and wide filled bars correspond to rectangular 90° and 180° pulses, respectively. Selective pulses and gradients are indicated as semi-ellipses. The default pulse phase is x. The proton carrier frequency is centred at the water frequency (5.7 ppm). The values for the 13C and 15N offsets are set to 77 ppm (13CRibose), 90 ppm (13CC1′), 70 ppm (13CC2′) and 160 ppm (15N), respectively. Asynchronous GARP decoupling (Shaka et al. 1985) is used to suppress heteronuclear scalar coupling during acquisition. The pulse field gradients with a length of 1 ms have a smoothed square amplitude (Bruker Topspin 2.0, 2006). They are applied along the z-axis and have the following strengths: G1:16%, G2:16% (pulse length 300 μs), G3:80%, G4:70%, G5:10%, G6:50%, G7:60%. 100% of gradient strength corresponds to 53.5 Gauss/cm. Fixed delays are adjusted as follows: Δ = 3 ms (1/(2*1JHC)), T = 6.26 ms (1/(4*1JCC). For the CC-TOCSY transfer the FLOPSY-16 mixing sequence (Kadkhodaie et al. 1991) was applied, optimized for a single transfer (τM = 3 ms) or multiple transfers (τM = 15 ms). For the virtual decoupling scheme, a band selective pulse 180° Q3 Gaussian cascade (Emsley and Bodenhausen 1992) of 2 ms (semi-ellipse) is applied either on C1′ or C2′. Phase cycling: φ1 = x, − x, φ2 = 2(x), 2(−x), φ IP3 = 4(y), 4(−y), φ AP3 = 4(x), 4(−x), φrec = x, 2(−x), x, −x, 2(x), −x. Quadrature detection in the ω1 and ω2 dimensions is obtained by incrementing φ1 and φ2 in a States-TPPI manner (Marion et al. 1989). The in-phase and anti-phase components of the C1′C2′ coherences were recorded in an interleaved manner and afterwards combined with a standard Bruker c-program (au-prog: splitcomb) using the parameter: ipap, inphase/antiphase correction term of 1.09 and an average coupling constant 1J(C1′C2′) = 40 Hz
In the new 3D (H)CC-TOCSY-H1′C1′ experiment (Fig. 2), proton magnetization is excited and the coherence is transferred via an INEPT step to carbon coherence. In-phase carbon coherence is generated in a second INEPT step with concomitant evolution of carbon chemical shift in a constant time period (1/J(C,C)). In the subsequent CC-TOCSY step using the mixing sequence FLOPSY-16 (Kadkhodaie et al. 1991), coherence is transferred to the C1′ carbon nuclei. In order to obtain the chemical shifts of the H1′ protons, a second transfer step generates transverse H1′ coherence and chemical shift evolves using an optimized spectral width of 2–3 ppm. We followed this strategy for the following reasons: (1) the H1′,C1′ spectral region reveals the best chemical shift dispersion within the ribose resonances in nucleic acids and (2) the resonance assignment of the H1′,C1′ spins can easily be correlated with the assignment of aromatic nuclei via HCN-type experiments (Sklenár et al. 1993) also for larger RNA constructs. The specific evolution of proton chemical shift after the TOCSY sequence might allow applying the pulse sequence for mixed protonated/deuterated RNAs that can be prepared biochemically (Batey et al. 1996; Vallurupalli et al. 2006; Lu et al. 2010) but can be omitted for uniformly deuterated RNAs. In order to obtain in-phase carbon coherence, the back transfer is concatenated with the application of an IPAP scheme, for which the 1J(C1′,C2′) carbon coupling has to be virtually decoupled only.
We recorded the 3D (H)CC-TOCSY-H1′C1′ experiment on a 14mer RNA with a mixing time of τM = 15 ms for the CC-spinlock in order to correlate all intraresidual ribose carbon nuclei as well as with a short mixing time of τM = 3 ms to exclusively detect the C2′–C1′ cross peak. The combined analysis of both experiments allows discrimination between the partially overlapping chemical shifts of C2′ and C3′ (Fig. 3). The related proton detected experiments, the HCCH-TOCSY (Kay et al. 1993) or the forward directed HCC-TOCSY-CCH-E.COSY (Schwalbe et al. 1995; Glaser et al. 1996; Marino et al. 1996), are typically performed using an RNA sample prepared in D2O, since some of the ribose 1H signals completely overlap with the H2O resonance frequency. In the new carbon direct detected NMR experiment, this problem is resolved and thus, also enables the use of RNA-samples in H2O.

2D strips out of the 3D (H)CC-TOCSY-H1′C1′ experiment for the 13C,15N-labelled 14mer RNA with a CC-TOCSY mixing period of τM = 15 ms (left) enabling magnetization transfer to all ribose carbons and a short mixing period of τM = 3 ms (right) to detect cross peaks originating from C1′ and C2′ only. Both experiments were recorded at a field strength of 14.4 T (600 MHz 1H frequency) at 298 K using a triple resonance cryogenic probe for 1H, 13C and 15N. The field strengths for 1H and 13C pulses were 22.7 and 20.3 kHz, respectively. During acquisition, GARP decoupling was applied at field strengths of 3.6 and 1 kHz for 1H and 15N, respectively. The FLOPSY mixing sequence was applied at a field strength of 8.3 kHz. Both 3D NMR experiments were recorded with 8 scans over a period of 17 h with 28, 28 and 1 k complex points in t1, t2 and t3. The acquisition time was set to 204 ms, t max2 was 28 ms and t max1 was 5.3 ms. A relaxation delay of 1 s was used
In addition, we applied the (H)CC-TOCSY-H1′C1′ pulse sequence to a 27mer RNA hairpin structure. This 27mer RNA is selectively 13C-labelled as indicated in Fig. 4 (specified in the “Materials and methods” section). Also here, the 3D experiment was recorded with two different mixing times, enabling different magnetization transfers. Both experiments were recorded on a 950 MHz spectrometer within a total time of 38 h.

Left Secondary structure of the 27mer RNA. Gray marked residues are 13C-labelled. 2D strips out of the 3D (H)CC-TOCSY-H1′C1′ experiment for the selectively 13C-labelled 27mer RNA with a CC-TOCSY mixing period of τM = 15 ms (middle) enabling magnetization transfer to all sugar carbons and a short mixing period of τM = 3 ms (right) to detect cross peaks originating from C1′ and C2′ only. Both experiments were recorded at a field strength of 22.3 T (950 MHz 1H frequency) at 298 K using a triple resonance cryogenic probe for 1H, 13C and 15N. The field strengths for 1H and 13C pulses were 19.6 and 23.8 kHz, respectively. During acquisition, GARP decoupling was applied at field strengths of 3.8 and 1 kHz for 1H and 15N, respectively. The FLOPSY mixing sequence was applied at a field strength of 8.3 kHz. Both 3D experiments were recorded with 8 scans for a period of 19 h with 28, 32 and 632 complex points in t1, t2 and t3. The acquisition time was set to 80 ms, t max2 was 32 ms and t max1 was 3.5 ms. A relaxation delay of 1 s was used
In addition to the resonance assignment itself, the here developed NMR experiments show a direct correlation of the sugar puckering and the sugar carbon chemical shift for both RNA hairpin structures (Ebrahimi et al. 2001; Fürtig et al. 2003). For the 14mer RNA, nucleotides U7 and C8 (Fig. 1) adopt C2′-endo conformation as derived from 3J(H1′,H2′) and 3J(H3′,H4′) homonuclear coupling constants (Schwalbe et al. 1994) and from dipole–dipole cross correlated relaxation rates \( \Upgamma_{{{\text{C1}}^\prime {\text{H1}},{\text{C2}}^\prime {\text{H2}}^\prime }}^{\text{c}} \) and \( \Upgamma_{{{\text{C3}}^\prime {\text{H3}}^\prime ,{\text{C4}}^\prime {\text{H4}}^\prime }}^{\text{c}} \) (Nozinovic et al. 2010a, b). A detailed analysis is beyond the scope of this manuscript. The 13C chemical shifts of U7 and C8, however, differ in comparison to the other nucleotides; especially C1′ and C4′ chemical shifts show a difference of 2–3 ppm and thus, qualitatively report on the ribose conformation.
For the unstructured heptaloop of the 27mer RNA, the ribose conformation has been determined by means of 3J(H1′,H2′) couplings and 13C heteronuclear NOE data (Fürtig et al. 2007). This analysis reported C3′-endo conformation for C11 and G15, whereas the ribose moieties of nucleotides G13, U14, U16, and U17 adopt C2′-endo conformation. The data for nucleotide U12 indicated conformational averaging. The qualitative analysis of the carbon chemical shifts observed in the 3D (H)CC-TOCSY-H1′C1′ experiment show that nucleotides G13 to U17 adopt C2′-endo conformation, and the conformational averaging of nucleotide U12 can also be inferred.
In addition, for the 27mer RNA we compared the (H)CC-TOCSY-H1′C1′ experiment with proton excitation (Fig. 2) with a shorter version of the experiment (CC-TOCSY-H1′C1′), utilizing direct carbon excitation with proton decoupling to build-up 13C-heteronuclear NOE enhancement during acquisition and relaxation delay (Aboul-ele et al. 1994). However, for the 27mer RNA, the experiment with proton excitation is a factor of 1.2–1.3 more sensitive than the experiment with the heteronuclear NOE build-up and carbon excitation.
The applicability of the described 13C-direct detected experiments to larger RNA structures is mainly limited by the sensitivity of the probe. This sensitivity can generally be increased using carbon-optimized cryogenic probes with a cold 13C-inner coil and a cold 13C-preamplifier which are commercially available but were not used in our case. We further demonstrate the general applicability of the experiment to larger RNA structures. Figure S1 shows the (H)CC-TOCSY-H1′C1′ experiment running on the 14mer RNA sample at 298 K in comparison with spectra recorded at 278 K. For lower temperatures, a reasonable 3D spectrum can be recorded by increasing the number of scans. In addition, we demonstrated the CC-TOCSY experiment on a 70mer RNA, the 2′-deoxyguanosine-dependent riboswitch RNA; results are shown in Figure S2. For the riboswitch RNA, the experiment starts with direct carbon excitation and for the detection, the IPAP step has been removed (for further details see Supporting Information). Both applications show that the 13C-direct detected experiments can be applied for larger RNAs. For this purpose additionally it is expected that further improvements can be obtained by using deuterated RNAs.
The (H)CPC-experiments for the sequential assignment of RNAs via the phosphodiester backbone
Virtual decoupling of 1J(C3′,C4′) and 1J(C4′,C5′)
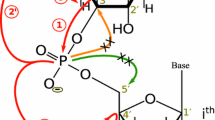
The 13C-direct detected (H)CPC-experiment (Figs. 6a, 7a) correlates the C4′ carbons with the adjacent 31P spins for a sequential assignment of RNAs via the phosphodiester backbone. This experiment is derived from the HCP experiment (Marino et al. 1994). In the 13C-direct detected (H)CPC-experiment, C4′ carbon coherence is excited after the initial INEPT step of the experiment and also detected; the (H)CPC experiment is therefore an out-and-back experiment (Kay et al. 1990). The C4′ spin of nucleotide i (C4′i) is coupled to two 31P spins, the 31Pi on the 5′-side and the 31Pi+1 on the 3′-side with 3J(C4′i,Pi) and 3J(C4′i,Pi+1) coupling constants of both around 8–10 Hz in standard A-form RNA helices. In addition, the C4′ spin is directly coupled to two carbon spins (C3′ and C5′) with 1J(C3′,C4′) ~ 38 Hz and 1J(C4′,C5′) ~ 42 Hz. In order to apply virtual decoupling during detection, we propose a new double IPAP scheme (Duma et al. 2003; Bermel et al. 2005) (Fig. 5). The 13C chemical shifts of the ribose moieties in RNA resonate over a relatively small range of chemical shifts of 35 ppm. Therefore, long selective pulses have been used e.g. to cover only the C4′ resonances (82 ± 3 ppm) corresponding to 6.5 ms for a RE-BURP (Geen and Freeman 1991) pulse. To selectively invert the C3′ and C4′ region simultaneously (77 ± 9.5 ppm), we used a 2 ms RE-BURP pulse. For selective refocusing of the C4′ (82 ± 3 ppm) and the C5′ (65 ± 3 ppm) carbons simultaneously, we used a band-selective-Bloch-Siegert-compensated RE-BURP pulse (Steffen et al. 2000) with a length of 6.5 ms. The double IPAP is applied in an interleaved manner and the linear combination of all four experiments (Fig. 5a) is performed after data acquisition by using the standard Bruker c-program splitcomb (included in Topspin 2.1). As shown in Fig. 5b, the four experiments might also be reduced to two, as the linear combination of the AP-AP spectrum and the IP-IP spectrum resulted also in one signal only. In this case, the pulse sequence might be shorter, but not all components are used for the linear combination and thus, the result is a factor of √2 less sensitive.

Double IPAP pulse scheme for RNA virtual decoupling of 1J(C3′,C4′) and 1J(C4′,C5′) illustrated on a 13C,15N-labelled UTP sample. The offset values for 13C are set to 77 ppm (13CRibose), 83.3 ppm (13CC4′), 69.4 ppm (13CC3′), and 64.7 ppm (13CC5′), respectively. The pulse field gradient of 1 ms length has a smoothed square amplitude (Bruker Topspin 2.0, 2006). It is applied along the z-axis and has the following strength: G1: 31%. 100% of gradient strength corresponds to 53.5 Gauss/cm. Fixed delays are adjusted as follows: T = 6.26 ms (1/(4*1JCC). φIPIP = x, −x, φIPAP = −y, y, φAPIP = −y, y, φAPAP = −x, x, φrec = −x, x. For the virtual decoupling, the following band selective pulses are applied: for C4′ (83 ppm ± 3 ppm) a 6.5 ms RE-BURP (Geen and Freeman 1991), for C3′ & C4′ (76 ppm ± 9.5 ppm) a 2 ms RE-BURP, for C4′ & C5′ (83 ppm ± 3 ppm & 64 ppm ± 3 ppm) a band-selective-Bloch-Siegert-compensated RE-BURP pulse with a length of 6.5 ms(RE-BURP_C4C5bs). The 1D spectrum a shows the linear combination of all four spectra using the c-prog splitcomb (included in Bruker Topspin 2.1). The doted lines indicate the 1D spectrum b, resulting from the linear combination of only the two spectra (IPIP and APAP)
The sequential assignment of the RNA backbone in the (H)CPC experiment
The new double IPAP scheme was incorporated in the carbon direct detected (H)CPC experiment (Fig. 6a). The coherence transfer pathway starts on the H4′ and magnetization is transferred via an INEPT step to C4′. Subsequently, coherence is further transferred to 31Pi and 31Pi+1 via 3J(C4′i,Pi) and 3J(C4′i,Pi+1), respectively.

Pulse sequences of a 2D-(H)CPC experiment, b 2D-HCP-CC-TOCSY-C1′ experiment and c 2D-(H)CPC-C5′ experiment with virtual decoupling in the acquisition dimension t2, respectively. Narrow and wide filled bars correspond to rectangular 90° and 180° pulses, respectively. Selective pulses and gradients are indicated as semi-ellipses. The default pulse phase is x. The proton carrier frequency is centred at the water frequency (4.7 ppm). The values for the 13C and 31P offsets are set to 77 ppm (13CRibose), 90 ppm (13CC1′), 70 ppm (13CC2′), 82 ppm (13CC4′) 60 ppm (13CC5′) and −0.8 ppm (31P), respectively. Asynchronous GARP decoupling (Shaka et al. 1985) is used to suppress heteronuclear scalar couplings during acquisition. The pulse field gradients of 1 ms length have a smoothed square amplitude (Bruker Topspin 2.0, 2006). They are applied along the z-axis and have the following strengths (experiment A): G1: 50%, G2: 30%, G3: 19%; (experiment B): G1: 50%, G2: 30%, G3: 19% G4: 80%, G5: 70%;(experiment C): G1: 50%, G2: 30%, G3:19% G4: 80%. 100% of gradient strength corresponds to 53.5 Gauss/cm. Fixed delays are adjusted as follows: Δ = 3 ms (1/(2*1JHC)), δ = 25 ms (1/(2*1JCP)), T = 6.26 ms (1/(4*1JCC). The FLOPSY-16 (Kadkhodaie et al. 1991) is used for the CC-TOCSY step with a τM = 32 ms. Phase cycling (experiment A): φ1 = x,−x, φ2 = 2(x), 2(−x), φ3 = 4(x), 4(−x), φ IPIP4 = 4(x), 4(−x), φ IPAP4 = 4(y), 4(−y), φ APIP4 = 4(y), 4(−y), φ APAP4 = 4(−x), 4(x), φrec = x,−x,−x,x, −x,x,x,−x. Phase cycling (experiment B): φ1 = x,−x, φ2 = 2(x), 2(−x), φ3 = 4(x), 4(−x), φ4 = 4(y), 4(−y), φ IP5 = 8(−y), 8(y), φ AP5 = 8(x), 8(−x), φrec = R, −R, −R, R, R = x, 2(−x), x. (experiment C):): φ1 = x,−x, φ2 = 2(x), 2(−x), φ IP3 = 4(−y), 4(y), φ AP3 = 4(−x), 4(x), φ IP4 = (x), φ AP4 = y, φrec = x,−x,−x,x, −x,x,x,−x. For all experiments, quadrature detection in the F1 dimension is obtained by incrementing φ2 in a States-TPPI manner (Marion et al. 1989). For the virtual decoupling, the following band selective pulses are applied: for C4′ (82 ppm ± 3 ppm) a 6.5 ms RE-BURP (Geen and Freeman 1991), for C3′ & C4′ (77 ppm ± 9.5 ppm) a 2 ms RE-BURP, for C4 & C5′ (82 ppm ± 3 ppm & 62 ppm ± 3 ppm) a band-selective-Bloch-Siegert-compensated RE-BURP pulse with a length of 6.5 ms and a C2′-band selective pulse 180° Q3 Gaussian cascade (Emsley and Bodenhausen 1992) of 2 ms (semi-ellipse)
In order to generate in-phase carbon coherence, the back transfer is concatenated with the double IPAP sequence to generate virtual decoupling of the homonuclear carbon–carbon couplings. Figure 7a schematically illustrates the magnetization transfer in the (H)CPC spectrum. To gain better resolution, the (H)CPC experiment can additionally comprise a CC-TOCSY step to transfer the sequential backbone walk in the well-resolved spectral region of the C1′ resonances. Such extension is similar to the HCP-CCH-TOCSY experiment originally developed using 1H-detection (Marino et al. 1995) (Fig. 6b). Additional sequential information can be obtained from the correlation of the C5′ carbons with the phosphorus nuclei. This was accomplished using the 2D-HCP-CC-TOCSY sequence (Fig. 6b) with a shorter mixing time of 6 ms for the CC-transfer. Alternatively, we developed a new pulse sequence with a COSY step interrelated in the virtual decoupling scheme (Fig. 6c). For generation of the correct in-phase or anti-phase magnetization, also the phase of the second last carbon 90 degree pulse was cycled. In fact, our results indicate that here, the COSY-transfer yields a better sensitivity compared to the TOCSY-transfer.

Upper left Schematic illustration of possible magnetization transfers via J(C,C) or J(C,P) couplings. a assigned 2D (H)CPC spectrum of the 14mer RNA. b assigned 2D (H)CP-CC-TOCSY-C1′ spectrum with a long CC-mixing time of τM = 32 ms for magnetization transfer to C1′. C: assigned 2D (H)CPC-C5′ spectrum with an additional transfer-step to C5′. All experiments were recorded on a 600 MHz NMR spectrometer and a temperature of 298 K using a cryogenic probe for 1H, 13C and 31P. The field strengths for 1H, 13C and 31P pulses were 23.8, 16.8 and 12.5 kHz, respectively. During acquisition, WALTZ and GARP decoupling sequences were applied at field strengths of 3.6 kHz and 833 Hz for 1H and 31P, respectively. The FLOPSY sequence was applied at a field strength of 8.3 kHz. The (H)CPC experiment was recorded with 512 scans for a period of 15 h. The (H)CP-CC-TOCSY-C1′ experiment was recorded with 1,792 scans for 52 h and the total experimental time for the (H)CPC-C5′ was 25 h. All three experiments were recorded with 20 and 1 k complex points in t1 and t2 by using a relaxation delay of 1 s. The acquisition time was set to 135 ms and t max1 was 24 ms for all three experiments
The results of the carbon-phosphorus correlated spectra, shown in Fig. 7, illustrate the general applicability of these experiments for the sequential assignment of nucleic acids. In the (H)CPC experiment with the C4′ to phosphorus correlations (Fig. 7a), the sequential walk along the loop and stem residues is illustrated. For the loop position C8, no coupling to the adjacent nucleotides could be detected, in agreement with previous results of the proton detected 3D HCP-experiment (Fürtig et al. 2003). Further information in order to complement and confirm the sequential assignment was obtained from the two additional experiments, the the (H)CP-CC-TOCSY-C1′ spectrum (Fig. 7b) that is based on the better resolved C1′ carbon-positions and the 2D (H)CPC-C5′spectrum (Fig. 7c) that connects the C5′carbons to phosphorus.
Conclusion
The current set of 13C-direct detected experiments allows a direct and unambiguous assignment of the majority of the hetero nuclei and the identification of the individual riboses and their sequential order for both, the 14mer and the 27mer RNA hairpin structures. Thus, the 13C-detected NMR methods are a useful complement to the traditional 1H-detected approach for the resonance assignment of oligonucleotides. Furthermore, these experiments are useful to quantitatively determine RNA backbone conformations based on 13C and 31P chemical shifts.
We show that 13C-direct detected experiments are also applicable to larger RNAs by optimizing hardware, sample preparation and pulse sequence. The sensitivity increased by using of carbon optimized probes with a cold inner carbon coil and a cold preamplifier. Further optimization of the pulse sequences is feasible by shortening delays or removing the virtual decoupling scheme. The 13C-direct detection experiment in combination with selectively deuterium-labelled RNAs, especially with only the H1′ being protonated, is expected to also yield the resonance assignment of the phosphodiester backbone moieties in larger RNA structures.
References
Aboul-ele F, Nikonowicz EP, Pardi A (1994) Distinguishing between duplex and hairpin forms of RNA by 15N–1H heteronuclear NMR. FEBS Lett 347:261–264
Andersson P, Weigelt J, Otting G (1998) Spin-state selection filters for the measurement of heteronuclear one-bond coupling constants. J Biomol NMR 12:435–441
Batey RT, Cloutier N, Mao H, Williamson JR (1996) Improved large scale culture of Methylophilus methylotrophus for 13C/15N labeling and random fractional deuteration of ribonucleotides. Nucleic Acids Res 24:4836–4837
Bermel W, Bertini I, Felli IC, Kümmerle R, Pierattelli R (2003) 13C direct detection experiments on the paramagnetic oxidized monomeric copper, zinc superoxide dismutase. J Am Chem Soc 125:16423–16429
Bermel W, Bertini I, Duma L, Felli IC, Emsley L, Pierattelli R, Vasos PR (2005) Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angew Chem Int Ed Engl 44:3089–3092
Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R (2006) C-13-detected protonless NMR spectroscopy of proteins in solution. Prog Nucl Magn Reson Spectrosc 48:25–45
Bertini I, Felli IC, Kümmerle R, Luchinat C, Pierattelli R (2004a) (13)C-(13)C NOESY: a constructive use of (13)C-(13)C spin-diffusion. J Biomol NMR 30:245–251
Bertini I, Felli IC, Kümmerle R, Moskau D, Pierattelli R (2004b) 13C–13C NOESY: an attractive alternative for studying large macromolecules. J Am Chem Soc 126:464–465
Cherepanov A, Glaubitz C, Schwalbe H (2010). High resolution studies of uniformly labeled RNA by solid-state NMR spectroscopy. Angew Chem (in revision)
Duma L, Hediger S, Lesage A, Emsley L (2003) Spin-state selection in solid-state NMR. J Magn Reson 164:187–195
Ebrahimi M, Rossi P, Rogers C, Harbison GS (2001) Dependence of 13C NMR chemical shifts on conformations of RNA nucleosides and nucleotides. J Magn Reson 150:1–9
Eletsky A, Moreira O, Kovacs H, Pervushin K (2003) A novel strategy for the assignment of side-chain resonances in completely deuterated large proteins using 13C spectroscopy. J Biomol NMR 26:167–179
Emsley L, Bodenhausen G (1992) Optimization of shaped selective pulses for NMR using a quaternion description of their overall propagators. J Magn Reson 97:135–148
Fares C, Amata I, Carlomagno T (2007) 13C-detection in RNA bases: revealing structure-chemical shift relationships. J Am Chem Soc 129:15814–15823
Felli IC, Pierattelli R, Glaser SJ, Luy B (2009) Relaxation-optimised Hartmann-Hahn transfer using a specifically Tailored MOCCA-XY16 mixing sequence for carbonyl-carbonyl correlation spectroscopy in 13C direct detection NMR experiments. J Biomol NMR 43:187–196
Fiala R, Sklenar V (2007) 13C-detected NMR experiments for measuring chemical shifts and coupling constants in nucleic acid bases. J Biomol NMR 39:153–163
Fürtig B, Richter C, Wöhnert J, Schwalbe H (2003) NMR spectroscopy of RNA. Chembiochem 4:936–962
Fürtig B, Richter C, Bermel W, Schwalbe H (2004) New NMR experiments for RNA nucleobase resonance assignment and chemical shift analysis of an RNA UUCG tetraloop. J Biomol NMR 28:69–79
Fürtig B, Wenter P, Reymond L, Richter C, Pitsch S, Schwalbe H (2007) Conformational dynamics of bistable RNAs studied by time-resolved NMR spectroscopy. J Am Chem Soc 129:16222–16229
Geen H, Freeman R (1991) Band-selective radiofrequency pulses. J Magn Reson 93:93–141
Glaser SJ, Schwalbe H, Marino JP, Griesinger C (1996) Directed TOCSY, a method for selection of directed correlations by optimal combinations of isotropic and longitudinal mixing. J Magn Reson B 112:160–180
Hennig M, Ott D, Schulte P, Lowe R, Krebs J, Vorherr T, Bermel W, Schwalbe H, Griesinger C (1997) Determination of homonuclear C-13-C-13J couplings between aliphatic carbon atoms in perdeuterated proteins. J Am Chem Soc 119:5055–5056
Hu KF, Vögeli B, Clore GM (2006) C-13-detected HN(CA)C and HMCMC experiments using a single methyl-reprotonated sample for unambiguous methyl resonance assignment. J Biomol NMR 36:259–266
Jordan JB, Kovacs H, Wang Y, Mobli M, Luo R, Anklin C, Hoch JC, Kriwacki RW (2006) Three-dimensional 13C-detected CH3-TOCSY using selectively protonated proteins: facile methyl resonance assignment and protein structure determination. J Am Chem Soc 128:9119–9128
Kadkhodaie M, Rivas O, Tan M, Mohebbi A, Shaka AJ (1991) Broad-band homonuclear cross polarization using flip-flop spectroscopy. J Magn Reson 91:437–443
Kay LE, Ikura M, Tschudin R, Bax A (1990) 3-dimensional triple-resonance NMR-spectroscopy of isotopically enriched proteins. J Magn Reson 89:496–514
Kay LE, Xu GY, Singer AU, Muhandiram DR, Formankay JD (1993) A gradient-enhanced Hcch Tocsy experiment for recording side-chain H-1 and C-13 correlations in H2o samples of proteins. J Magn Reson Ser B 101:333–337
Kim JN, Roth A, Breaker RR (2007) Guanine riboswitch variants from Mesoplasma florum selectively recognize 2′-deoxyguanosine. Proc Natl Acad Sci U S A 104:16092–16097
Kovacs H, Moskau D, Spraul M (2005) Cryogenically cooled probes—a leap in NMR technology. Prog Nucl Magn Reson Spectrosc 46:131–155
Lu K, Miyazaki Y, Summers MF (2010) Isotope labeling strategies for NMR studies of RNA. J Biomol NMR 46:113–125
Marino JP, Schwalbe H, Anklin C, Bermel W, Crothers DM, Griesinger C (1994) A 3-dimensional triple-resonance H-1, C-13, P-31 experiment—sequential through-bond correlation of ribose protons and intervening phosphorus along the RNA oligonucleotide backbone. J Am Chem Soc 116:6472–6473
Marino JP, Schwalbe H, Anklin C, Bermel W, Crothers DM, Griesinger C (1995) Sequential correlation of anomeric ribose protons and intervening phosphorus in RNA oligonucleotides by a 1H, 13C, 31P triple resonance experiment: HCP-CCH-TOCSY. J Biomol NMR 5:87–92
Marino JP, Schwalbe H, Glaser SJ, Griesinger C (1996) Determination of gamma and stereospecific assignment of H5′ protons by measurement of (2)J and (3)J coupling constants in uniformly C-13 labeled RNA. J Am Chem Soc 118:4388–4395
Marion D, Ikura M, Tschudin R, Bax AJ (1989) Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J Magn Reson 85:393–399
Meissner A, Duus JO, Sorensen OW (1997) Integration of spin-state-selective excitation into 2D NMR correlation experiments with the heteronuclear ZQ/2Q pi rotations for 1JXH- resolved E.COSY-type measurements of heteronuclear coupling constants in proteins. J Biomol NMR 10:89–94
Nozinovic S, Fürtig B, Jonker HR, Richter C, Schwalbe H (2010a) High-resolution NMR structure of an RNA model system: the 14-mer cUUCGg tetraloop hairpin RNA. Nucleic Acids Res 38:683–694
Nozinovic S, Richter C, Rinnenthal J, Jonker H, Fürtig B, Schwalbe H (2010b). Quantitative 2D and 3D gamma-HCP experiments for the determination of the angles alpha and zeta in the phosphodiester backbone of oligonucleotides. J Am Chem Soc (in press)
Ohlenschläger O, Haumann S, Ramachandran R, Görlach M (2008) Conformational signatures of 13C chemical shifts in RNA ribose. J Biomol NMR 42:139–142
Ottiger M, Delaglio F, Bax A (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson 131:373–378
Pervushin K, Eletsky A (2003) A new strategy for backbone resonance assignment in large proteins using a MQ-HACACO experiment. J Biomol NMR 25:147–152
Quant S, Wechselberger RW, Wolter MA, Wörner KH, Schell P, Engels JW, Griesinger C, Schwalbe H (1994) Chemical synthesis of C-13-labeled monomers for the solid-phase and template controlled enzymatic-synthesis of DNA and RNA oligomers. Tetrahedron Lett 35:6649–6652
Richter C, Reif B, Griesinger C, Schwalbe H (2000) NMR spectroscopic determination of angles alpha and delta in RNA from CH-dipolar coupling, P-CSA cross-correlated relaxation. J Am Chem Soc 122:12728–12731
Rinnenthal J, Richter C, Nozinovic S, Fürtig B, Lopez JJ, Glaubitz C, Schwalbe H (2009) RNA phosphodiester backbone dynamics of a perdeuterated cUUCGg tetraloop RNA from phosphorus-31 NMR relaxation analysis. J Biomol NMR 45:143–155
Schwalbe H, Marino JP, King GC, Wechselberger R, Bermel W, Griesinger C (1994) Determination of a complete set of coupling constants in 13C-labeled oligonucleotides. J Biomol NMR 4:631–644
Schwalbe H, Marino JP, Glaser SJ, Griesinger C (1995) Measurement of H, H-Coupling Constants Associated with Nu-1, Nu-2, and Nu-3 in Uniformly C-13-Labeled RNA by HCC-TOCSY-CCH-E.Cosy. J Am Chem Soc 117:7251–7252
Serber Z, Richter C, Moskau D, Bohlen JM, Gerfin T, Marek D, Haberli M, Baselgia L, Laukien F, Stern AS, Hoch JC, Dötsch V (2000) New carbon-detected protein NMR experiments using CryoProbes. J Am Chem Soc 122:3554–3555
Serber Z, Richter C, Dötsch V (2001) Carbon-detected NMR experiments to investigate structure and dynamics of biological macromolecules. Chembiochem 2:247–251
Shaka AJ, Barker PB, Freeman RJ (1985) Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson 64:547–552
Shimba N, Stern AS, Craik CS, Hoch JC, Dötsch V (2003) Elimination of 13Calpha splitting in protein NMR spectra by deconvolution with maximum entropy reconstruction. J Am Chem Soc 125:2382–2383
Shimba N, Kovacs H, Stern AS, Nomura AM, Shimada I, Hoch JC, Craik CS, Dötsch V (2004) Optimization of 13C direct detection NMR methods. J Biomol NMR 30:175–179
Sklenár V, Peterson RD, Rejante MR, Feigon J (1993) Two- amd three-dimensional HCN experiments for correlating base and sugar resonances in 15N, 13C-labeled RNA oligonucleotides. J Biomol NMR 3:721–727
Steffen M, Vandersypen LMK, Chuang IL (2000) Simultaneous soft pulses applied at nearby frequencies. J Magn Reson 146:369–374
Stoldt M, Wöhnert J, Görlach M, Brown LR (1998) The NMR structure of Escherichia coli ribosomal protein L25 shows homology to general stress proteins and glutaminyl-tRNA synthetases. EMBO J 17:6377–6384
Vallurupalli P, Scott L, Hennig M, Williamson JR, Kay LE (2006) New RNA labeling methods offer dramatic sensitivity enhancements in 2H NMR relaxation spectra. J Am Chem Soc 128:9346–9347
Vögeli B, Kovacs H, Pervushin K (2005) Simultaneous 1H- or 2H-, 15N- and multiple-band-selective 13C-decoupling during acquisition in 13C-detected experiments with proteins and oligonucleotides. J Biomol NMR 31:1–9
Wenter P, Reymond L, Auweter SD, Allain FH, Pitsch S (2006) Short, synthetic and selectively 13C-labeled RNA sequences for the NMR structure determination of protein-RNA complexes. Nucleic Acids Res 34:e79
Wijmenga SS, van Buuren BNM (1998) The use of NMR methods for conformational studies of nucleic acids. Prog Nucl Magn Reson Spectrosc 32:287–387
Acknowledgments
Financial support by the Access to Research Infrastructures activity in the 6th Framework Programme of the EC (Contract # RII3-026145, EU-NMR) for conducting the research is gratefully acknowledged. Harald Schwalbe is member of the DFG-funded Cluster of Excellence: macromolecular complexes.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Richter, C., Kovacs, H., Buck, J. et al. 13C-direct detected NMR experiments for the sequential J-based resonance assignment of RNA oligonucleotides. J Biomol NMR 47, 259–269 (2010). https://doi.org/10.1007/s10858-010-9429-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-010-9429-5